Hello
We are simulating a system with many rigid bodies. Is there a way to ask LAMMPS to print out the principle axes for every rigid body in the system (at t=0, for example)? (without editing the LAMMPS code?)
Priiority: This is not critical for us. If LAMMPS does not have this feature, we have another way to get around our problem. But we are curious.
Thank you very much in advance.
Andrew
----- details (feel free to skip) -----
We can compute the principal axes ourselves, but there is ambiguity about the principal axis polarities. Even if we sort by eigenvalues, and consider only right-handed coordinate systems, there are a number of choices (4) for the principal axis of any rigid body. Example:
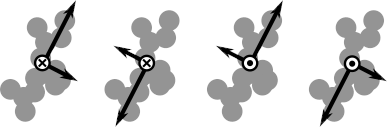
Is there a way we can ask LAMMPS which choice it used for each rigid body in our system? (More details below…)
— why we want this information (feel free to skip) —
My coworker and I are running simulations with millions of identical copies of the same molecule (each with a different molecule-ID) which we constrain to be rigid (currently using “fix rigid/small molecule”). My coworker wants to reduce the size of the trajectory files by only saving the quaternions for each rigid body in the dump file (instead of all of the atom coordinates). He is using these commands to do this, and it seems to be working:
fix fxRigid gRigid rigid/small molecule
compute 1 all rigid/local fxRigid mol xu yu zu quati quatj quatk quatw#xu yu zu quati quatj quatk quatw
dump 2 all local 100 tmp.dump index c_1[1] c_1[2] c_1[3] c_1[4] c_1[5] c_1[6] c_1[7] c_1[8]
My coworker has written a viewer which can read these trajectory files, but he has questions about how to interpret the quaternions. We know that each quaternion (c_1[5] c_1[6] c_1[7] c_1[8]) stores the orientation of the principal axes of each molecule (relative to the global world coordinate system). And we can compute the principal axis of these molecules ourselves (at t=0, for example). But we don’t know which of the polarity choices LAMMPS is choosing (see picture above), so when we apply the quaternions to our molecules, we might end up with 75% of our molecules inverted (but otherwise oriented correctly).
We know how to modify the LAMMPS code to print out this information, but that won’t help other people who use our code (unless we tell them how to modify the LAMMPS code too).
Our solution so far was to copy the LAMMPS code which computes the principal axes (from molecule.cpp) into our own viewer code which computes principal axes. This way, the two codes will agree on the axis polarity. This works fine for us, but if there’s another better way to do this, please let us know.