Dear Everyone,
I have a question on the way that Lammps calculates the total (molecular) potential energy for the system including Improper group.
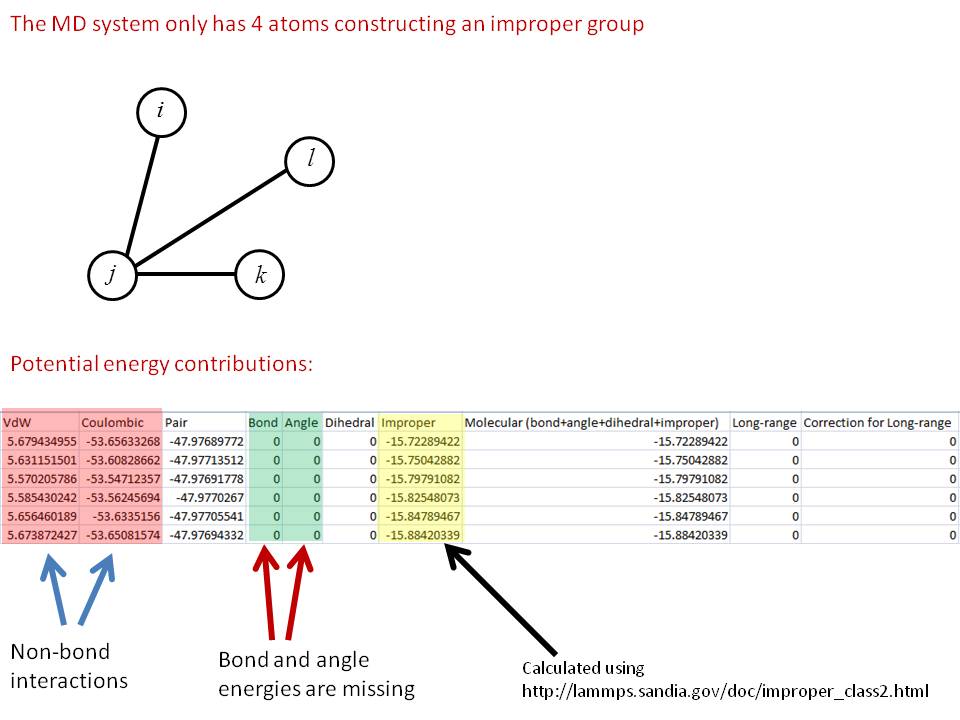
I use Class2 potential, and my MD system only has 4 atoms constructing an improper group (please see the figure attached)
The individual contribution of the total potential energy is also shown.
Is there an error in the code to calculate such total (molecular) potential energy, since there are three bond energy and three angle energy missing in the total potential energy.
This is very different than the way Lammps calculates the potential energy for system including dihedral group.
Thank you very much for your comments.
Lili Zhang