Respected Sir,
Thank you so much for your guidance.
Kind Regards,
Safikul
Respected Sir,
Thank you so much for your guidance.
Kind Regards,
Safikul
Dear Sir,
I did according to your suggestion. I am getting following error. It is not reading forces from vasprun.xml file. How can I rectify this?
Also Sir, I have two questions:
I have many vasprun.xml files for each rattled_supercell structures. Sir, how can I add all these files to the python script. You have suggested to add only one rattled_supercell structure.
As per the error Sir I think I have to add DFT forces to the rattled_supercell structures to get modified rattled_supercell structures which contain positions as well as forces of each atom.
Sir, please help me.
Kind Regards.
Safikul
Error:
File “/home/safikul_akd/Anaconda_physics/lib/python3.9/site-packages/hiphive/utilities.py”, line 56, in _get_forces_from_atoms
if atoms.calc is not None and calc is not None:
AttributeError: ‘Atom’ object has no attribute ‘calc’
Print your atoms object and check how they look like. It sounds like you gave the prepare_structure a Atom object and not an Atoms object.
If you want to add multiple structures you can just make a simple loop, if you can add one structure to the StructureContainer it should not be a problem to add multiple.
Dear Sir,
By the use of the command supercell_rattled=read('vasprun.xml') it is reading only the positions and forces of the atoms. But it is unable to find displacements of atoms. For StructureContainer function I am getting the following error. How can I add displacements of each atom to the supercells_rattled structures?
Kind Regards,
Safikul
Error
File “/home/safikul_akd/Anaconda_physics/lib/python3.9/site-packages/hiphive/newone.py”, line 29, in
sc.add_structure(structures)
File “/home/safikul_akd/Anaconda_physics/lib/python3.9/site-packages/hiphive/structure_container.py”, line 165, in add_structure
if ‘displacements’ not in atoms_copy.arrays.keys():
AttributeError: ‘list’ object has no attribute ‘arrays’
As I wrote above you can use supercell_training = prepare_structure(supercell_rattled, supercell_ideal).
Sir, I think I have not framed my questions properly. My questions are:
In supercell.rattled.extxyz file each row contains 7 terms. The first 3 terms for displacements of atoms generated by Monte Carlo Simulations and the next 3 terms for the atomic force components acted on each atom. What is the last term?
In supercell.rattled.extxyz files the generated supercells has different type of POSCAR format (example-Sn S Sn S Sn S) than VASP POSCAR format(Sn S). I have not used ASE as VASP interface. I have changed the POSCAR format of the supercells generated by hiphive structure generation to the POSCAR format of VASP and have done the DFT calculations by VASP . Then I have changed the Supercell_ideal POSCAR format generated by hiphive to VASP POSCAR format. When I am adding the Supercell_ideal and Supercell_rattled structures to the scripts it takes only displacements and force components. But the 7th term it does take.
Then I am getting the above error. Sir, please provide your valuable suggestions.
Kind Regards,
Safikul
You dont need manually to convert any VASP output files to .extxyz files. ASE can directly read the output files from VASP. Format Specific Options — ASE documentation
Dear Sir,
I am generating POSCAR_supercell= prim.repeat((2,2,1)). Here POSCAR_supercell looks like as follows. But this format gives me error when I am calculating energies as here positions of the atoms are Sn S Sn S Sn S Sn S
2 2 2 2 2 2 2 2
but the correct POSCAR format is Sn S and it gives no error.
8 8
Same thing happens when I am generating the supercell rattled structures by the function generate_mc_rattled_structures the POSCAR format changes.
Sir, then how can I generate supercell_rattled and supercell_ideal with correct POSCAR format? This is my actual question.
Kind Regards,
Safikul
POSCAR_supercell
Sn S Sn S Sn S Sn S
1.0000000000000000
8.1623043781240501 0.0000000000000000 0.0000000000000000
0.0000000000000000 8.5675465877304529 0.0000000000000000
0.0000000000000000 0.0000000000000000 20.4505126865094518
Sn S Sn S Sn S Sn S
2 2 2 2 2 2 2 2
Cartesian
1.0543207008577737 1.6847006093933385 7.3632398021480059
3.1504265094592712 3.8175035139584033 10.1529149864768371
3.0874041778490020 4.1259029676178622 7.5341635269122538
1.0257183820151239 1.9620645463483479 9.8557947590321522
1.0497140985096942 5.9410619659955444 7.2639449601307051
3.0609151382839457 8.0468977974556068 10.1775706621598570
3.1013641220746879 8.5158704725166743 7.6665986438756768
0.9298188947139155 6.3210790498523730 9.9807508023115048
5.2027949392299133 1.6884148011810745 7.2547145288367698
7.1901296192382471 3.8447000519767482 10.1335358910843354
7.1235756914059332 4.1748413748228712 7.6330392822733151
5.1528142962064125 1.9399970476685104 9.8458178551551612
5.1152623231256165 5.9545184808542384 7.1844822992372173
7.1585023097575773 8.0277336089202738 10.2841321423076053
7.1305407437143637 8.4626003552260549 7.6662558327610082
5.1098722948808506 6.2392884237744930 9.9905617131413571
Dear Sir,
Please forgive me I am asking questions again and again. Sir, I am getting the following error. I have added all the vasprun.xml files. Again this error comes. The script and the error are as follows. Sir, please tell me what I should do now.
Kind Regards,
Safikul
Script
“”"
Construct a ForceConstantPotential from training data generated previously.
Runs in approximately 100 seconds on an Intel Core i5-4670K CPU.
“”"
import ase.io
from ase.io import read,write
from ase.io import vasp
from hiphive import ClusterSpace, StructureContainer, ForceConstantPotential
from hiphive.utilities import prepare_structures,get_displacements
from trainstation import Optimizer
from ase.calculators.vasp import Vasp
from ase.io.vasp import read_vasp,write_vasp
prim = read_vasp(‘prim’)
atom_ideal = read_vasp(‘supercell_ideal’)
zero=ase.io.read(filename=‘vasprun.xml0’,index=-1,format=‘vasp-xml’)
one=ase.io.read(filename=‘vasprun.xml1’,index=-1,format=‘vasp-xml’)
two=ase.io.read(filename=‘vasprun.xml2’,index=-1,format=‘vasp-xml’)
three=ase.io.read(filename=‘vasprun.xml3’,index=-1,format=‘vasp-xml’)
four=ase.io.read(filename=‘vasprun.xml4’,index=-1,format=‘vasp-xml’)
five=ase.io.read(filename=‘vasprun.xml5’,index=-1,format=‘vasp-xml’)
six=ase.io.read(filename=‘vasprun.xml6’,index=-1,format=‘vasp-xml’)
seven=ase.io.read(filename=‘vasprun.xml7’,index=-1,format=‘vasp-xml’)
eight=ase.io.read(filename=‘vasprun.xml8’,index=-1,format=‘vasp-xml’)
nine=ase.io.read(filename=‘vasprun.xml9’,index=-1,format=‘vasp-xml’)
value=[zero,one,two,three,four,five,six,seven,eight,nine]
print(value)
cutoffs = [8.0, 3.0]
cs = ClusterSpace(prim, cutoffs)
print(cs)
cs.print_orbits()
for atoms in value:
structures = prepare_structures(value, atom_ideal)
sc = StructureContainer(cs)
for structure in structures:
sc.add_structure(structures)
print(sc)
opt = Optimizer(sc.get_fit_data())
opt.train()
print(opt)
fcp = ForceConstantPotential(cs, opt.parameters)
fcp.write(‘SnS.fcp’)
print(fcp)
Error
File “/home/safikul_akd/Anaconda_physics/lib/python3.9/site-packages/hiphive/structure_container.py”, line 165, in add_structure
if ‘displacements’ not in atoms_copy.arrays.keys():
AttributeError: ‘list’ object has no attribute ‘arrays’
Respected Sir,
Thank you so much for your guidance so far. I am so grateful. It is working now. Sir, I have one query and that is how many rattle structures should I generate for proper calculation if I want to calculate for 5x5x1 supercell.
Kind Regards,
Safikul
For harmonic phonons i think its probably enough to use ~5 structures. And I would recommend using standard rattle with rattle_std=0.01, and not MC-rattle
Respected Sir,
I have determined the phonon dispersion curve of SnS monolayer with number of rattle_supercell structures equal to 10, cutoffs=[10.0,3.0]. After enforcing rotational sum rules the phonon dispersion looks fine. All the negative frequencies are gone. And the lattice thermal conductivity I have found~10 W/mK at temperature=300K which almost matches with the value =16 W/mK published in papers. The material has anharmonic bonds. When I am applying strain to the material keeping all the parameters same then the ZA branch has more negative frequencies. Then I am applying rotational sum rules with varying alpha parameters. But the phonon dispersion curves are not modifying with varying alpha parameters for strained structures. So, my questions are:
How many rattle structures should I use if the material has anharmonic bonds?
The next question is why after enforcing rotational sum rules the unstrained structure ZA branch is getting corrected with varying alpha parameters but the strained structure ZA branch is not getting corrected with varying alpha parameters.
Sir, please clarify these issues.
Kind Regards,
Safikul
alpha_vals = np.logspace(-10, 10, 50)
Respected Sir,
I am trying to enforce rotational sum rules to the structure SnS monolayer with 6% biaxial strain having 10 rattle supercells. The high symmetry path is (Gamma-X-S-Y-Gamma). It has negative frequencies near two Gamma points for the acoustic branches. I am varying alpha over wide range. The dispersion curves are getting modified for small and large alpha values. But only the negative frequencies near right hand side Gamma point are getting modified and giving linear behavior. Sir, my questions are:
How can I justify that a structure is unstable because applying rotational sum rules the negative frequencies are getting modified?
The negative frequencies near right hand Gamma point are getting linear behavior after enforcing rotational sum rules but the negative frequencies near left hand Gamma point are not getting modified and still shows negative behavior after enforcing rotational sum rules although I am varying alpha over wide range. Why is this happening?
Sir, I am humbly requesting to you to please answer these questions.
Kind Regards,
Safikul
Im not sure I understand your questions, maybe some figures of your obtained phonon dispersions would be helpful?
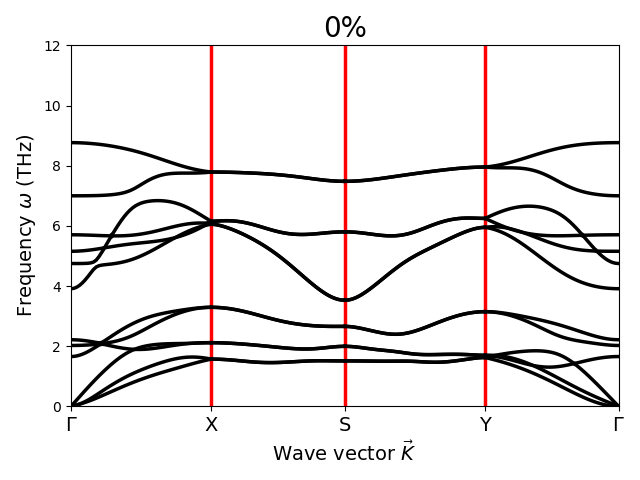
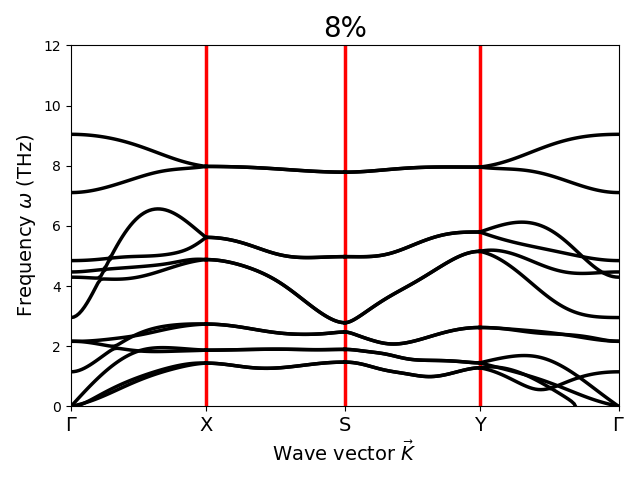
Respected Sir,
I have applied strain to the material. These phonon dispersion curves are obtained after enforcing rotational sum rules. For 6% strain case negative frequencies near Gamma point are appearing. I have varied alpha values over a wide range. Still negative frequencies are coming. So, my question is:
Kind Regards,
Safikul
I dont know much about the material or system, but yes it is possible that the instability is real but could also be an artifact due to fcs construction.
Maybe someone in science talk knows more about your material and what happens when strained etc.
Thank you so much Sir.
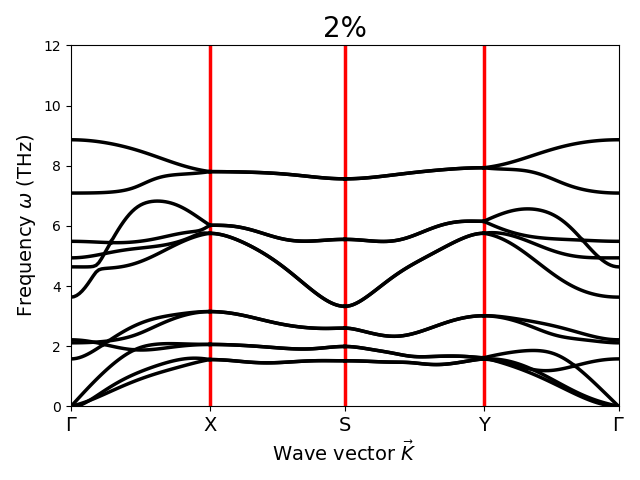
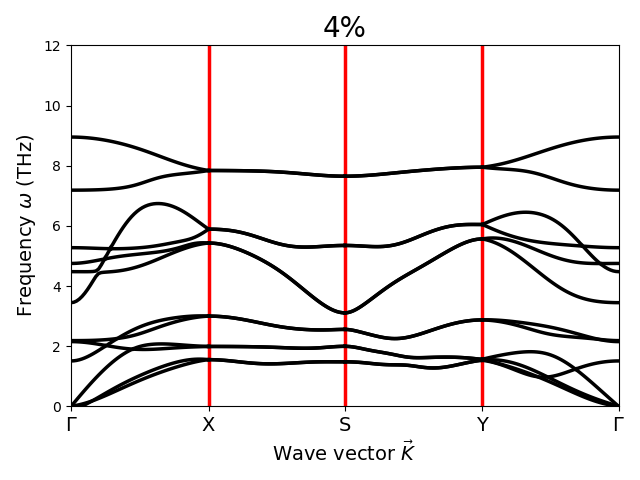
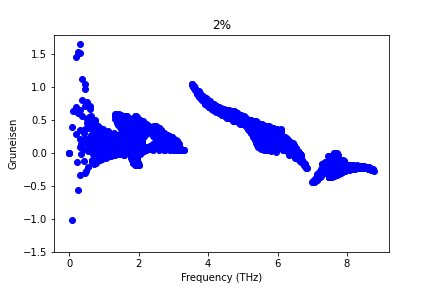
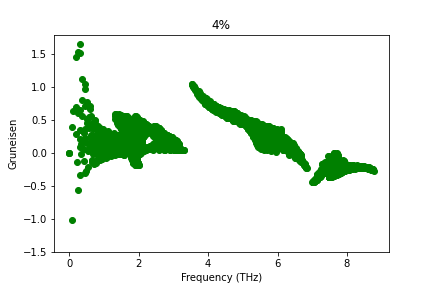
Dear Sir,
I have calculated Gruneisen parameter with Phono3py for different strained structure of SnS monolayer with the FC3 obtained by hiphive. But Gruneisen parameter does not change. What could be the possible reason for this? Sir, please suggest.
Kind Regards,
Safikul