Hello,
I’m running some simulations of benzene in a flexible porous framework. I have two groups of atoms, those of the adsorbate and those of the framework. I use separate time integration fixes for these two groups (since the benzene molecules are rigid), and then I set velocities (zeroing out linear momentum) and run.
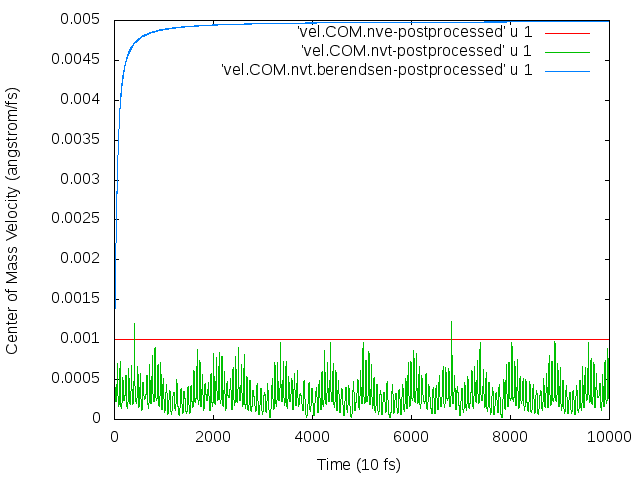
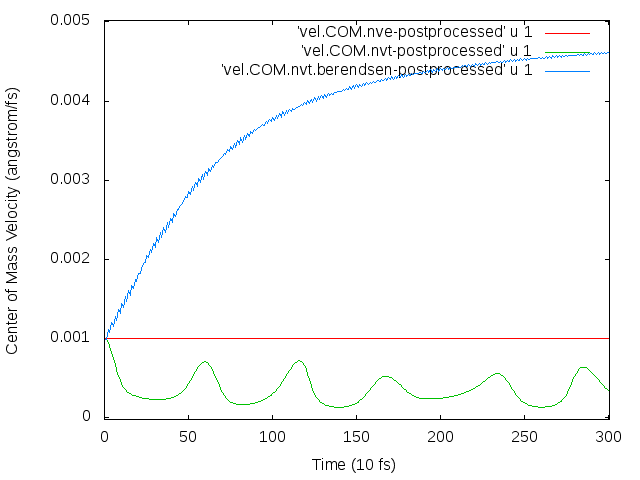
The atoms then begin to drift continuously in a particular direction. I’m trying to figure out why this drift occurs, what I need to do to fix it, and whether I need to remove the velocity bias before thermostatting to the temperature I want.
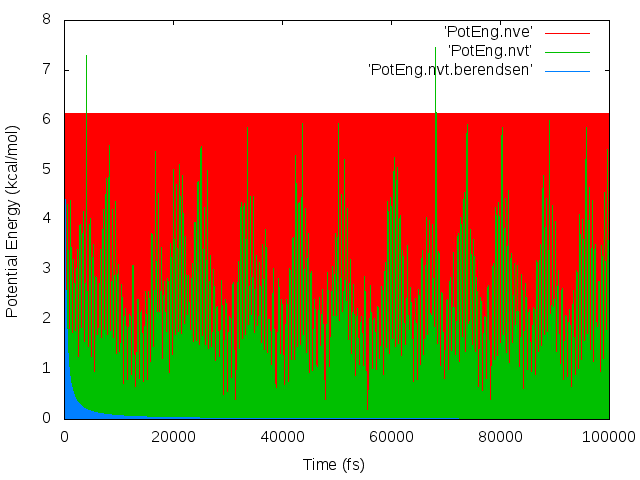
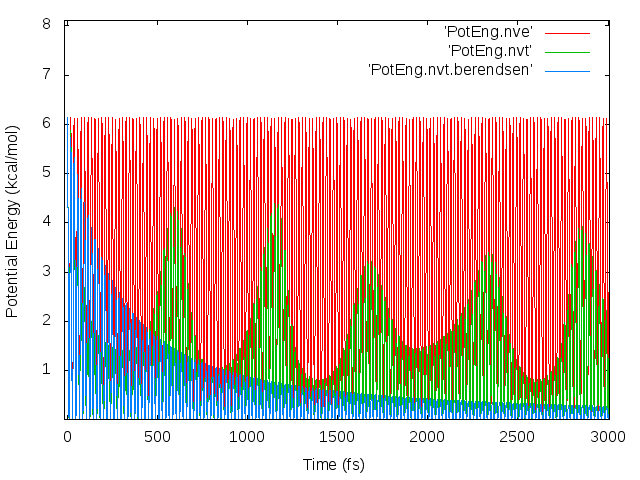
The thermostat is what’s causing the drift. When I ran the simulation in the NVE ensemble, no drift occurred. (Note that energy in the NVE ensemble was conserved, so the timestep is not too large, and drift occurs in the NVT ensemble even when I lowered the timestep from 0.5 to 0.1 fs, in response to the suggestion of http://lammps.sandia.gov/threads/msg34724.html.)
I suspect that the problem is due to the flying ice cube effect, though I’m not completely sure. My working through the solutions proposed in http://lammps.sandia.gov/threads/msg53543.html:
- Timestep: tested (see above), not the issue.
- Cutoff: The cutoff of 12.0 that I’m using is almost as large as it can be with a 25.8 Angstrom box, and it’s also more than 3X bigger than the greatest LJ sigma, so I can’t imagine this is the issue.
- Kspace convergence: I turned off long-range electrostatics, and the drift still occurs.
Note that when I have just the benzene atoms in the system, or when I have just framework atoms in the system, no drift occurs. It’s only when both are there that I see drift. Why would the flying ice cube effect only be present if the full system is in place, but not when each half is there separately?
Finally, if it is the flying ice cube effect, I’m not quite sure how to fix it since my simulation has rigid bodies. Per http://lammps.sandia.gov/threads/msg56336.html, “fix momentum” would be the thing to use, but per http://lammps.sandia.gov/threads/msg34731.html, “fix momentum” doesn’t work when rigid bodies are present. I’m guessing that “set velocity zero linear” has the same problem. Any way to zero out the linear momentum with rigid molecules?
A minimal working example is below, with the data file attached. Any pointers would be greatly appreciated.
Efrem Braun
#settings
units real
atom_style full
neigh_modify delay 0 every 1
pair_style lj/cut/coul/long 12.0
pair_modify mix arithmetic shift yes
special_bonds lj/coul 0.0 0.0 1.0
kspace_style pppm 1.0e-6
bond_style harmonic
angle_style harmonic
dihedral_style harmonic
improper_style cvff
#initialize atomic positions and FF
read_data “data_init.data”
#settings
group Benzene type 1 5
group MOF type 2 3 4 6 7 8 9
timestep 0.5
thermo 200
dump 1 all custom 200 traj.dump id mol type x y z ix iy iz xu yu zu
#run
fix 1 Benzene rigid/nvt molecule temp 300 300 100.0
fix 2 MOF nvt temp 300 300 100.0
run 0
velocity all create 300 38564
run 400000
data_init.data (348 KB)