Please reply to the list.
Your first set (lattice diamond 5.465 orient x 1 -1 0 orient y 0 0 1 orient z 1 1 0 origin 0.0 0.0 0.0) does not conform to the right-hand rule.
LAMMPS (5 Nov 2016)
3d Lennard-Jones melt
units lj
atom_style atomic
lattice diamond 5.465 orient x 1 -1 0 orient y 0 0 1 orient z 1 1 0 origin 0.0 0.0 0.0
ERROR: Lattice orient vectors are not right-handed (…/lattice.cpp:235)
Your second one does. How did you generate the same results with two different commands in which one clearly returns with an error?
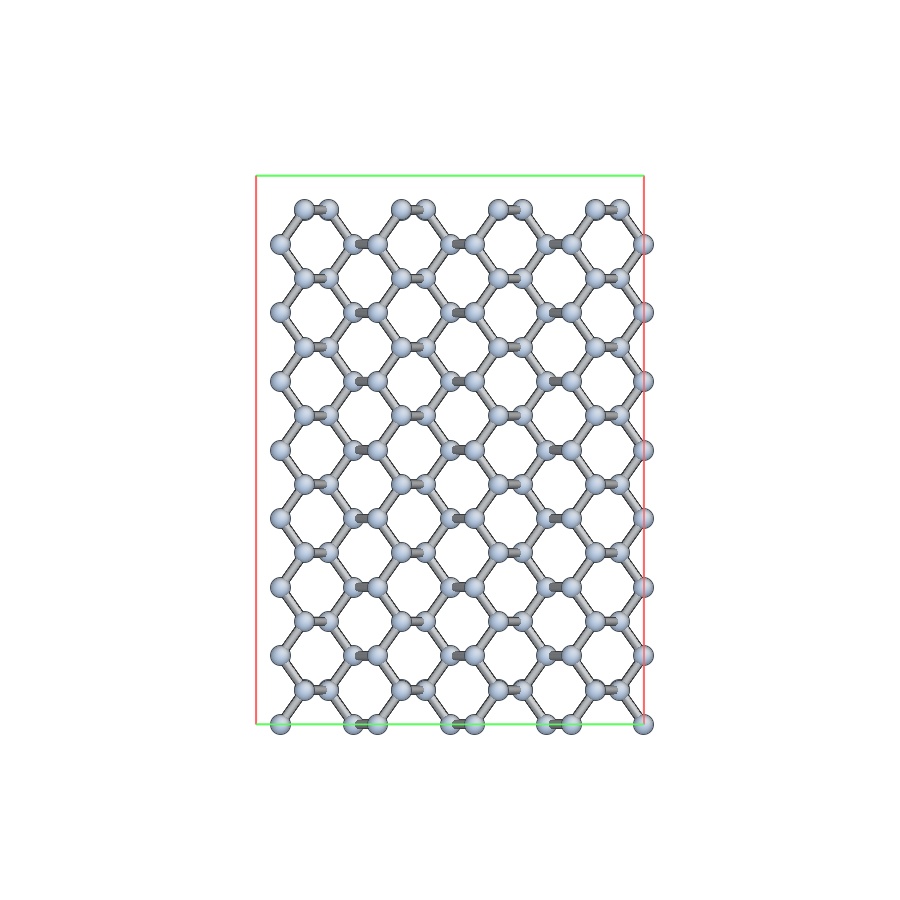
The second one does generate a good lattice with (110) facing X – see attached snapshot.
From your snapshots it is not clear what are the crystallographic orientations these axes are aligned to. It may be you are just visualizing different directions. The likelihood with LAMMPS’ lattice orientation having a bug is extremely low.
Ray
Actually, you are right about the command which does not conform the right-hand rule since I type it as wrong in the previous email.
Wrong one is:
orient x 1 -1 0 orient y 0 0 1 orient z 1 1 0 origin 0.0 1.0 0.0
Correct one I tried in Lammps:
orient x 0 0 1 orient y 1 -1 0 orient z 1 1 0 origin 0.0 1.0 0.0
I’m sorry for wrong typing to the e-mail (there is the correct one in my lammps input,)
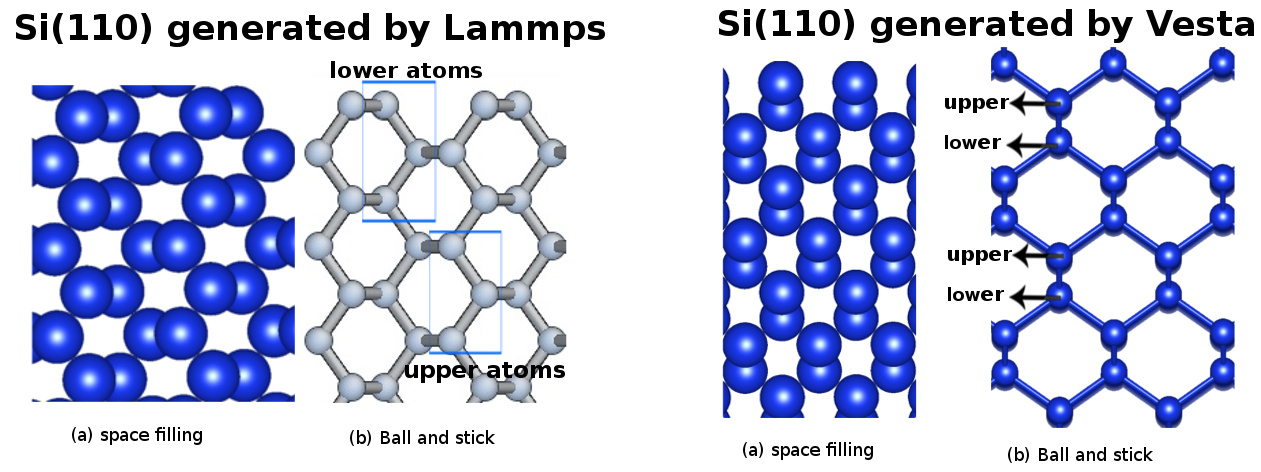
Sorry for repeating the same question but I still get the same problem, I prepared more detailed png which includes the Si(110) generated by both Lammps (on your image) and vesta and indicated the difference between two images.
Also I got the same image in two version: (a) Space filling (where the atomic arrangements are more clearly seen) (b) Ball and stick.
Would you like to check the image file, I provide more detail with it.
Best,
Nadire
No problem – glad we sorted one thing out.
The (110) plane normal from LAMMPS is along x (the perpendicular direction in your snapshot), while it seems from vesta the (110) plane normal is – again in your snapshot – horizontal. They are actually identical if you look from the same directions.
There is still no compeliing proof that LAMMPS is wrong. Have you tried radial distribution fuction, which would be the compelling proof?
Ray