Dear Lammps users,
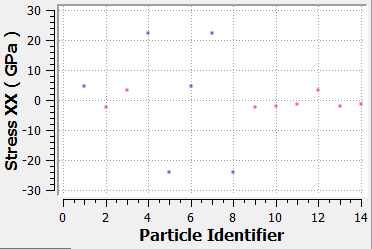
Attached you can see the atomic stress distribution of my ceramic system ( Stress-XX ) on my single lattice structure in its minimized positioned. This stress has been calculated using the following code:
compute stress all stress/atom NULL
dump q2 all custom 1 dump.npt.*.cfg id type xs ys zs c_stress[1] c_stress[2] c_stress[3] c_stress[4] c_stress[5] c_stress[6]
As you can see some atoms experiences high value of stress. I’m searching for a probable explanation. I don’t think anything is wrong with my potential or structure because it gives me correct elastic constants, lattice constants and P.E. value. So, does it make sense to you ? For the information, I used two different potential. But, both of them end with the same distribution. Moreover, when I built a large system and minimized and equilibrate it I observed the same behavior. However, I should mention that the distribution of the all component of the stress follows Gaussian distribution with center on 0 which is a nice thing I guess.
Thanks,
Ali