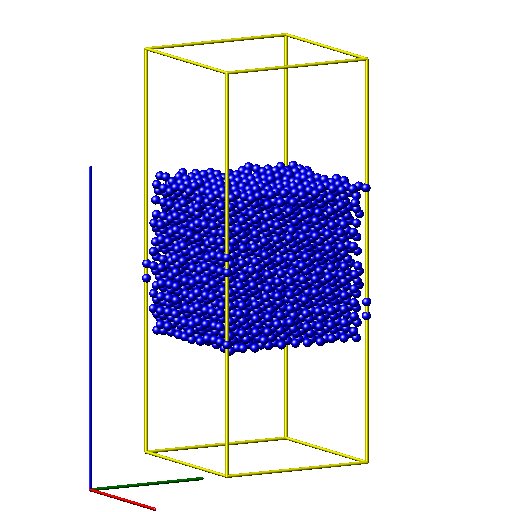
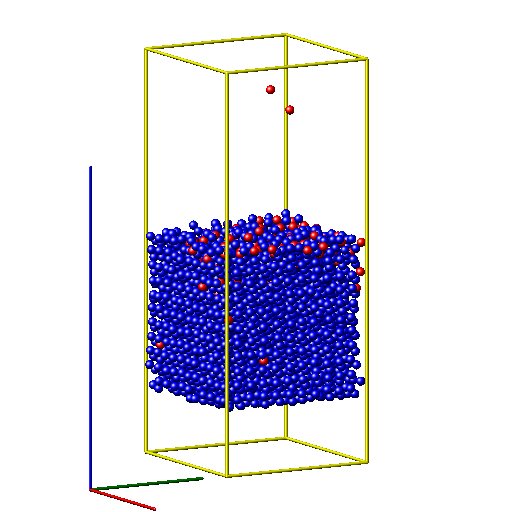
Dear Lammps users,
I am calculating metal substrate with incident oxygen chemical reaction problem with ReaxFF method. The substrate is at the center of the box and two vaccum slabs are positioned in the upper and bottom of the substrate respectively. After a long run, my substrate drifted down some distances from its original position. How can I avoid such thing?
Dear Lammps users,
I am calculating metal substrate with incident oxygen chemical reaction
problem with ReaxFF method. The substrate is at the center of the box and
two vaccum slabs are positioned in the upper and bottom of the substrate
respectively. After a long run, my substrate drifted down some distances
from its original position. How can I avoid such thing?
add one more layer of atoms at the bottom of your slab and immobilize
them (e.g. do not include them in time integration). this is common
practice for slab geometries. please see previous discussions on slab
geometry simulations in the mailing list archives for additional
details.