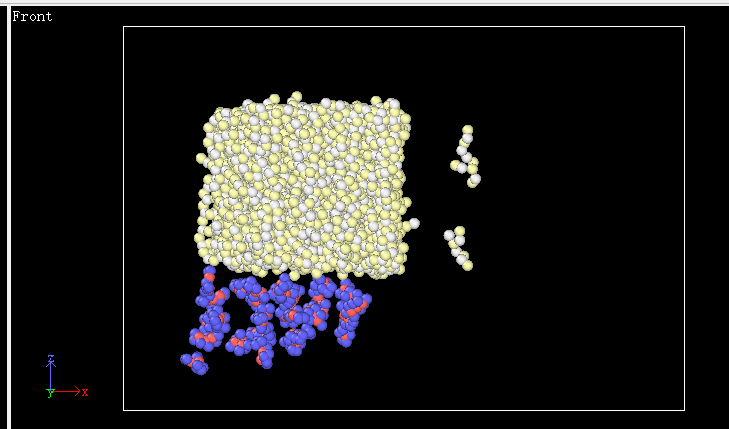
Dear All,
I tried to relax the system of silicon nitride and PTFE(polymer with the elements of C and F).unfortunately ,while the system is relaxing , the atoms belonging to Si3N4 is running apart as shown below. And the PTFE was supposed to be adsorbed to the Si3N4 surface ,but it didn’t.
it is strange for me because the temperature I set is not high and the potential is fine to my system I guess.
Below is my input file. Hopefully some one can shed light on this problem.
Thank you in advance
Yours sincerely
Tina
units real
dimension 3
boundary p p f
neighbor 0.8 bin
atom_style full
neigh_modify delay 5 one 5000 page 1000000
pair_style hybrid tersoff lj/cut/coul/long 12
bond_style class2
read_data PTFEpackmol.data extra/atom/types 2
read_data si3n4command.data add append offset 2 0 0 0 0 group si3n4 shift -10 -17 20
pair_coeff * * tersoff Si3N4.tersoff NULL NULL N Si
pair_coeff 1 1 lj/cut/coul/long 0.054 4.01
pair_coeff 2 2 lj/cut/coul/long 0.059 3.2
pair_coeff 1 2 lj/cut/coul/long 0.056 3.61
pair_coeff 2 4 lj/cut/coul/long 0.107 3.825
pair_coeff 2 3 lj/cut/coul/long 0.0623 3.635
pair_coeff 1 3 lj/cut/coul/long 0.059 4.4
pair_coeff 1 4 lj/cut/coul/long 0.101 4.23
kspace_style ewald 1.0e-5
kspace_modify slab 3.0
region ptfe block INF INF INF INF INF 20
group ptfe region ptfe
compute tep all temp
velocity all create 300 987098 temp tep
fix 1 all nvt temp 300 300 100
fix 2 all temp/rescale 100 300 300 0.01 1
fix 3 si3n4 setforce 0 0 0
timestep 0.1
thermo 100
dump 1 all custom 100 nvtxyz.lammpstrj id type x y z
run 18000