Dear all,
I used LAMMPS to perform NPT simulations of bilayer graphene oxide in water.
The water model is TIP4P/2005 (Abascal, J. L. F. & Vega, C. The Journal of Chemical Physics 123, 234505-234512 (2005).)
Graphene edges is functionalized by hydroxyl groups. The initial coordinates of chemical groups are taken from previous DFT study (Yan, J.-A. & Chou, M. Y. Physical Review B 82, 125403 (2010).). The force field for graphene oxide I use in MD simulations is OPLS-AA.
The time step is 0.1 fs. The total simulation time is 100 ps. The boundaries in three directions is all periodic.
The ensemble is NPT of 298K thermostat and 1atm isotropic barostat.
(fix 4 all npt temp 298.0 298.0 100.0 iso 1.0 1.0 1000.0)
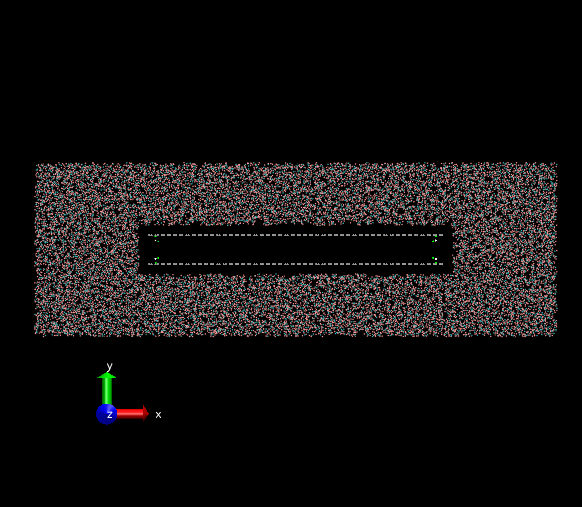
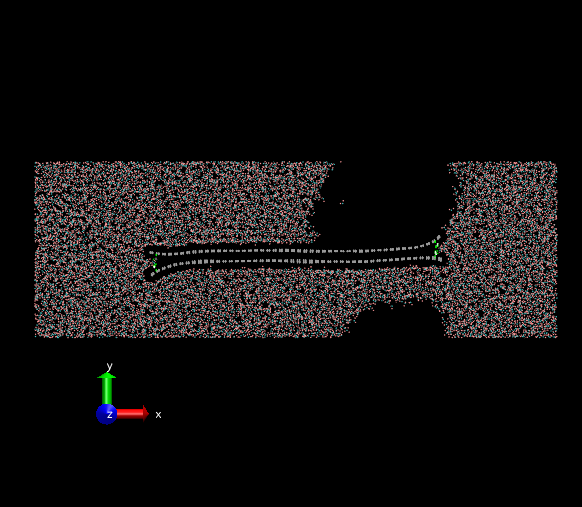
The initial state is a non-equilibrium configuration. The two layers of graphene oxide tend to be stuck together. The remaining space should be filled by surrounding water molecules. However, a large vacuum hole emerges and the simulation box won’t contract its size to decrease the vacuum hole. The attached PNG images are the initial and final frames of my simulation. The formation of the vacuum hole seems to be unrealistic. Is there anyone have the same problems in non-equilibrium simulations within water molecules? I am wondering how to resolve this issue.
I found a paper (Patel, R. Y. & Balaji, P. V. The Journal of Physical Chemistry B 109, 14667-14674 (2005)) raise a question for different MD results of isotropic and anisotropic pressure coupling methods. I can change my pressure coupling method from “iso” to “aniso”, which the xyz pressures are “couple none”. (fix 4 all npt temp 298.0 298.0 100.0 aniso 1.0 1.0 1000.0). Is it the key to solve this problem?
Thank you in advance.
Best,
-Wenpeng
The input script and log file of my simulation are attached as below:
########### input script ##########
System and potential definition
units real
boundary p p p
atom_style full
processors 2 2 2
read_data ./data.GO-WM
Pair Coeffs