I am using BKS potential to generate amorphous structures of Silicon oxide.
According to lammps mailing list, I am using Paul Crozier’s table (cf. lammps user’s list:). Thanks to Paul Crozier sharing that useful table. I first tried to reproduce the calculations from the original BKS paper on alpha quartz (PRL 64, 16, 1990). I built the initial structure with vmd and I also tried with vesta. I built 3 differents data file, 2 of them in a triclinic box with an xy shift (1x1x1 unit and 2x2x2 units) and the other one in an orthogonal box (2x2x2 units). See data files attached. Here is the lattice parameters and the cii elastic constants I found (I attached the input files): BKS 1x1x1 unit triclinic: a=5.079 A, c=5.590 A, C11=155.5 GPa and C33=188.4 GPa ; BKS 2x2x2 units triclinic: a=5.082 A, c=5.587 A, C11=206.9 GPa ; BKS 2x2x2 units ortho: C11=213.5 GPa ; PRL: a=4.941 A, c=5.449 A, C11=90.5 GPa and C33=107.0 GPa ; (I also tried with reaxFF: a=4.920 A, c=5.631 A and C11=96.7 GPa). As you can see, my BKS calculations over estimate the elastic constant by a factor of 2. reaxFF give me results similar to the experiment. I tried to parameterised buck/coul/long pair style with the coefficients in the PRL but my results are worst. Does anyone have an idea about that? Thank you, Nicolas
Sounds like you’ve already tested that SiO2 table in greater detail than I have. I’ve not calculated elastic constants, etc. with this force field, so I don’t have useful comments other than suggest you contact the PRL authors and ask them for input. Or perhaps someone else on this list will have suggestions for you.
While I didn't look it up to refresh my memory, I am pretty sure BKS uses charged particles with long range coulomb. And now that I followed the trace of messages I see that Pauls Tables include the short range part of coulomb to be used with ewald compatible long range like this:
Christian,
I cannot use pppm style with a triclinic box and that is why I built an alternative orthorhombic box in order to use the kspace commands.
Results on the elastic constants are about the same order of magnitude (two times greater than in PRL paper).
Carlos,
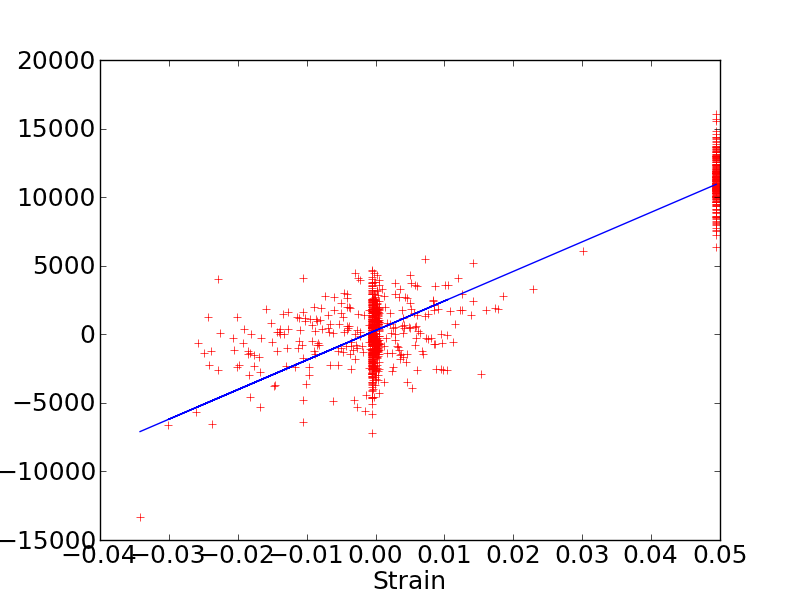
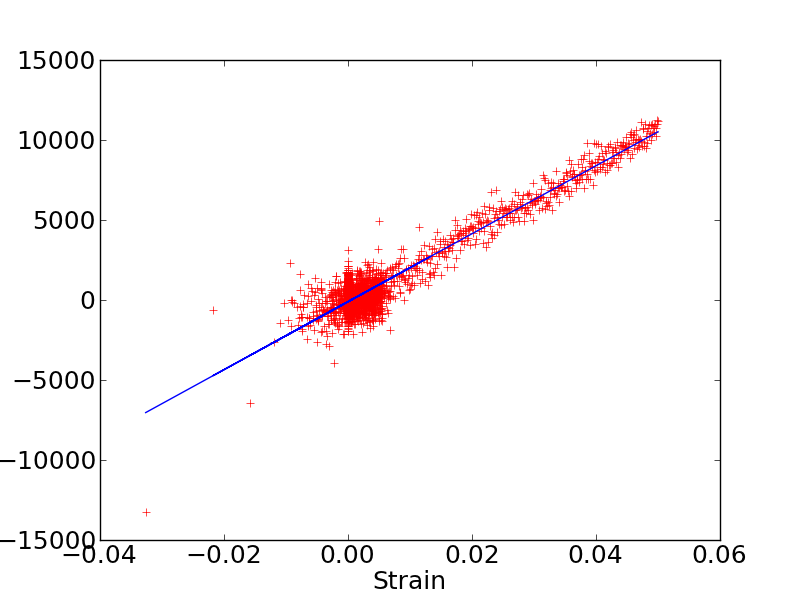
I just tried to use the change_box command as proposed in lammps user guide to compute elastic constants. I attach the stress (MPa)/strain plot. My C11 elastic constant is still around 200 GPa.
you are still doing it wrong then. You can not use the table values without pppm or ewald. In essence you are trying to use a screened Coulomb potential which is not what BKS did. What you could do is either generating new table files which use a simple coulomb cutoff potential or use buck/coul/cut with parameters calculated from what BKS did. In either case you need to use a fairly large cutoff then, I guess something in the range of 20-30A should be ok to average out enough over a SiO2 crystal.
Dear all,
As I said before, pppm is not available for triclinic boxes BUT I built a 2x2x2 units system in an orthorhombic box (data file attached) in order to use (input file attached):
Nicolas,
You better check your data file. The 2x2x2 cell you attached is wrong.
Which units are you using?
Certainly not Angs or Bohr as there you have a cell volume of ~1000
(Angs^3 or Bohr^3) and 72 atoms.
This give you a Volume_per_atom ~ 13.9 (Angs^3 or Bohr^3). In quartz
the Volume_per_atom ~ 55 Angs^3.
See: http://database.iem.ac.ru/mincryst/s_carta.php?QUARTZ+3895
And then you also have atoms outside the box with negative coordinates, etc.
The problem is all on your side 'cause as I said: Quartz has been
studied so many times that
Prob_of_original_paper_having_a_mistake -> 0.0
Carlos
Sorry, Carlos, but I think Nicolas's structure is correct, at least in
terms of volume per atom. The reference you provided gives a
volume/atom of 12.55 angs^3, which Nicolas's quartz is reasonably
close to.
Also, coordinates outside the box get wrapped around the periodic
boundary - no problem with that either.
Hi Nicolas,
If you are still having issues with the BKS elastic constants you
could try computing them from measuring the fluctuations of the
pressure tensor instead. This might help you to better understand
where the observed discrepancies come from.
Carlos