Thank you for your attention.
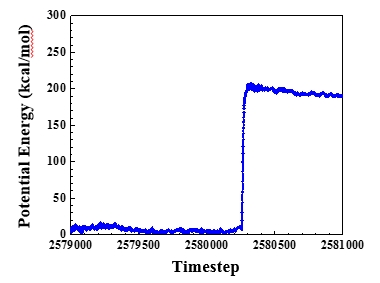
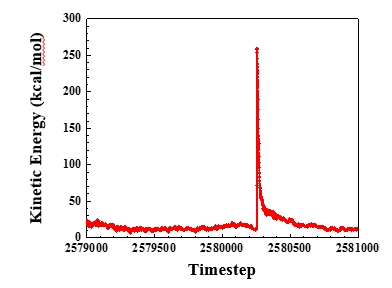
I came up with the dihedral potential parameters myself for fitting the dihedral angle distribution of supperatoms from coarse-grained simulation to one from all-atom simulation. But the coarse-grained simulations with dihedral potential always fail.
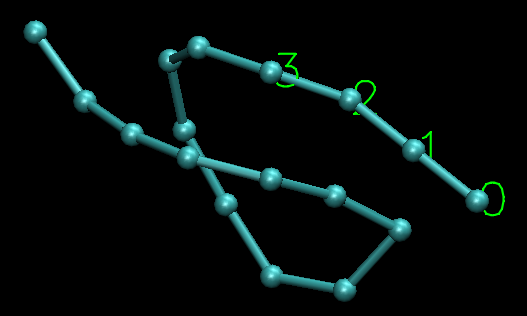
I observed the configuration(the picture is attached) of the coarse-grained chain when the bonded atoms would be far apart at the next timestep and found that the angle of three neighborest atoms is 179.9954 degree(near to 180 degree). I think it may be this angle(near to 180 degree) resulting in the bad dynamics. When the angle is very close to 180 degree, the distance between atom 0 and bond formed by atom 1 and atom 2 is so short(only 0.000377 Angstrom) that the force from the torque resulted from dihedral potential to atome 0 becomes very large. And then the atom 0 will fly away. So is it right that the dihedral potential can not be used when the angles are able to be close to 180 degree? Above it is my explaination. However, the fact is that the atom 1 flew away. It is so werid. I do not understand the fundamental algorithms about lammps well, so it is just my thought from the physcial reasoning.