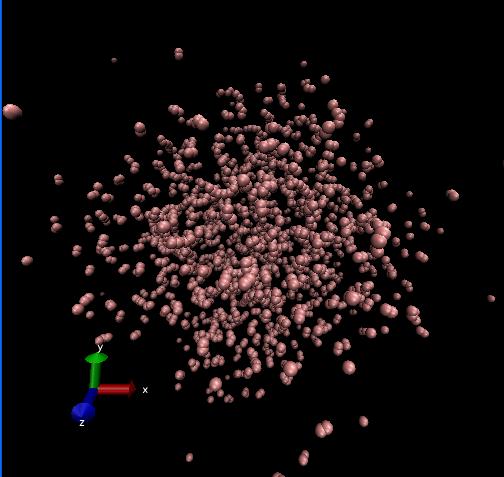
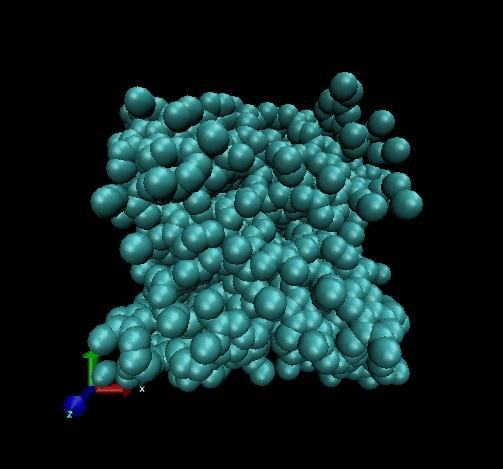
I have a problem about Carbon atoms system(there are bond carbon atoms and non-bond carbon atoms in this system).
I use tersoff potential and npt ensemble for getting stable structure, but the atoms in this the system scat at beginning, I don't know why?
Meanwhile I use DL_POLY to do the same job by using tersoff potential, it seem OK and it can get to the stable state, Actually, I wanna use lammps for later simulation. Can you help me about this problem.
thanks so much!
(in file and two photos of result (Lammps and DL_POLY separately) are attached in this letter)
ps:
# tersoff carbon
units metal
dimension 3
boundary p p p
atom_style atomic
read_data data.carbon
pair_style tersoff
pair_coeff * * SiC.tersoff C
compute mobile all temp
velocity all create 300.0 482748 temp mobile
fix 2 all npt temp 300.0 300.0 1.0 iso 0.0 0.0 100.0
compute pe all pe/atom
timestep 0.003
thermo 100
dump 1 all atom 100 dump.lammpstrj
dump 4a all custom 100 dump.data c_pe
run 1000
I have a problem about Carbon atoms system(there are bond carbon atoms and
non-bond carbon atoms in this system).
I use tersoff potential and npt ensemble for getting stable structure, but
the atoms in this the system scat at beginning, I don't know why?
Meanwhile I use DL_POLY to do the same job by using tersoff potential, it
are you using the same tersoff parameterization for carbon in both codes?
there are at least four different parameter sets for carbon in the
lammps distribution...
Thanks so much for your great help!
I exactly check the parameters of tersoff potential in both lammps and dlpoly, there are two differences (R and D).
I also change ensemble to nve with fix temp/rescale, it seem work better than dlpoly, the system quickly come into stable state, no atom scatting. Now I have a question why npt with thermostat and barostat can’t make the system stable (almost all the atom will scat)?
Meanwhile, can I get some specific IT infrastructure data from Splunk and use it in lammps?
Thanks a lot again! On 2011-6-30 15:44, Steve Plimpton wrote:
Thanks so much for your great help!
I exactly check the parameters of tersoff potential in both lammps and
dlpoly, there are two differences (R and D).
I also change ensemble to nve with fix temp/rescale, it seem work better
than dlpoly, the system quickly come into stable state, no atom scatting.
well, the fix temp/rescale still does modify the system's
kinetic energy. if that does make the difference you describe
it means that your original configuration is of very high
potential energy and that you first need to minimize it.
Now I have a question why npt with thermostat and barostat can't make the
system stable (almost all the atom will scat)?
NPT is to be used for a system that is close to equilibrium and
is not meant to remove a large amount of kinetic energy quickly.
in any case, it is good practice to run some minimization first
before running dynamics, if you initial structure is of high
potential energy (which will cause the instability you observe).
Meanwhile, can I get some specific IT infrastructure data from Splunk and
use it in lammps?
no idea. it is the sourceforge marketing crap that comes with
having them host a mailing list for free.