Hi,
I’m trying to simulate a membrane model using LAMMPS.
This triangulated membrane model consists of beads connected through hard bonds. Hard repulsion at close range is employed to prevent bead overlapping. There is a harmonic dihedral associated with the two neighboring triangles of each bond to simulate the bending energy of the membrane. The above three are all the interactions in this membrane model.
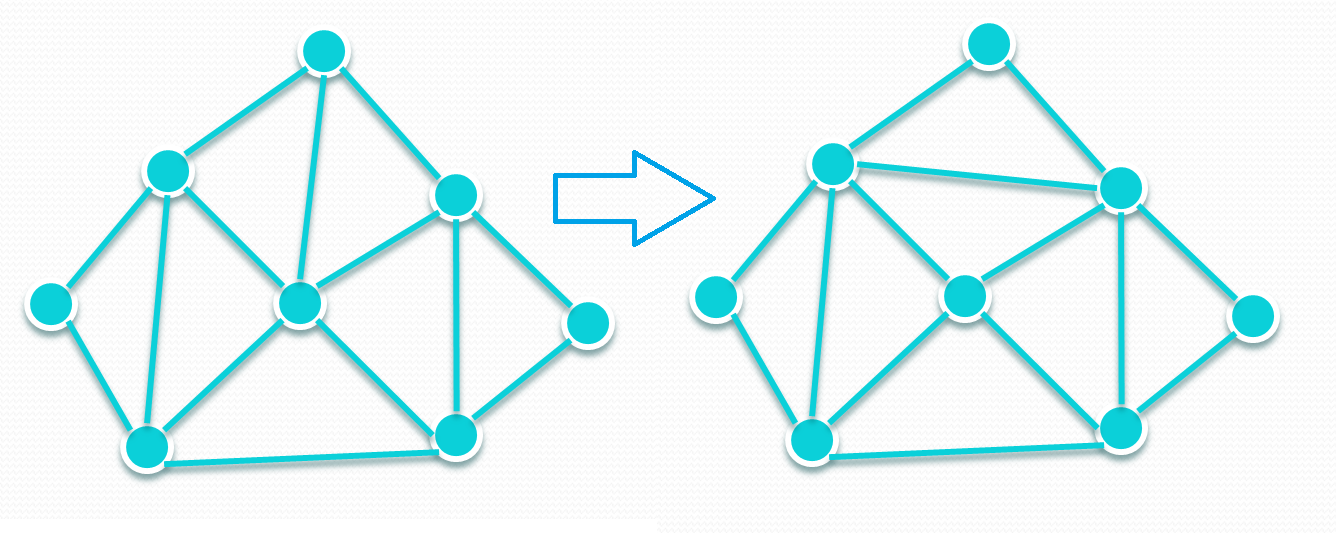
What’s special is, to add fluidity into this model, bond flipping needs to be performed. Please have a look at the attached image, which demonstrates one bond flipping. Each bond flipping requires changing the data of one bond and several related dihedrals. Every N time steps, the entire membrane needs to be scanned, and all eligible bonds need to be flipped.
This model is described in this paper: http://iopscience.iop.org/0295-5075/12/4/002
I suppose LAMMPS can’t do the above bond flipping, right? So I’m thinking about coding a new fix for this. I have some experience in C/C++ but I’ve never modified LAMMPS. I read the manual, developer guide, and dived into the source code. I think I’m clear about the data structure and how to change bonds, dihedrals. However, to flip a bond near a processor boundary, several processors are involved and have to work together, because a processor can only change its owned bonds, dihedrals (correct?), and a bond flipping requires changing all neighboring dihedrals that can possibly cross the processor boundary. So this requires communication among processors, which I don’t quite understand. I vaguely know I need to implement the pack_comm, unpack_comm, pack_reverse_comm, unpack_reverse_comm methods, and possibly pack_exchange and unpack_exchange?
What’s the difference between forward communication and reverse communication?
I’m really lost in the complexity of LAMMPS. Maybe what I have thought so far is entirely garbage. Please point me to the right direction.
Any help, suggestion, advice, insight is welcome.
Thanks!
Sincerely,