Dear LAMMPS forum,
I have:
- two slabs of gamma-alumina (43.68 * 26.27 * 21.06Å; LWH) immersed in water
- initial separation in z of 25Å, with 30Å thickness of water between all other sides and the periodic box boundaries
- Total simulation box size: 45 * 86.27 * 127.12Å (x, y, z)
I am trying to pull the upper slab down towards the lower one, to a separation of 3Å, in increments of 1Å - the objective is eventually to extract the PMF using WHAM.
For the moment I’ve run quite a quick simulation, just to get an idea of what happens:
- 10k steps of 0.1fs, followed by 150k fs (1fs timestep) of just letting the system run.
- This is followed by (what should be) moving 22Å in increments of 1Å, spending 100k fs at each stage.
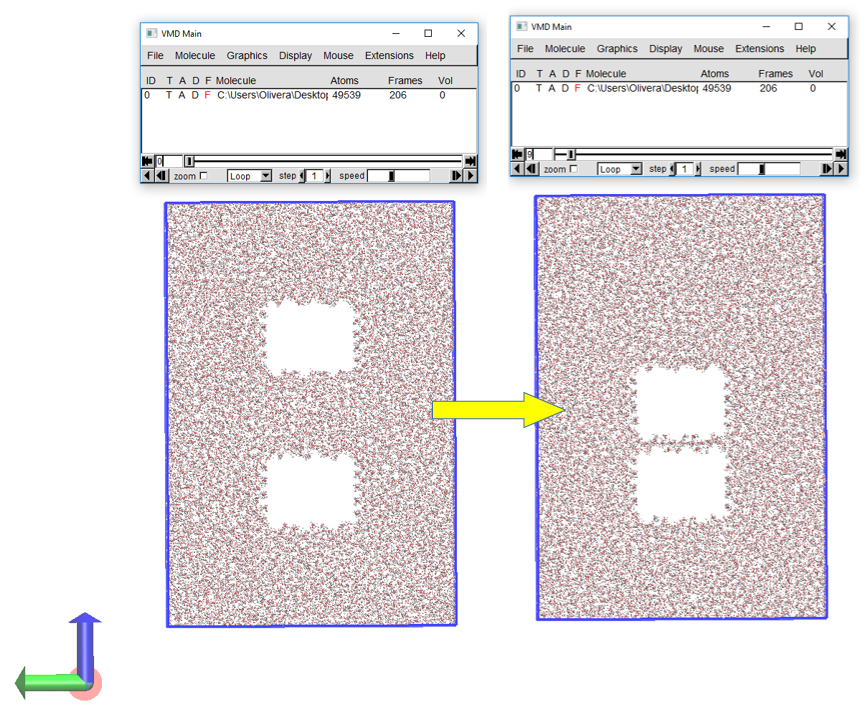
According to my .o file this seems to be happening (not able to attach in this message - 900kb attachment size limit). However, when I visualise the ouput.data file in VMD** it appears that this distance is closed in 9 frames (so ~90-100fs) which seems ridiculously fast.
**Attached screenshot composite from VMD: ‘transition.png’ (I’ve omitted the slab atoms for clarity)
I would be hugely grateful if anyone has any ideas on what’s possibly happening and why there seems to be this discrepancy. I’ve attached my run and .colvars files, if this helps.
I suspect it might be that I’ve made a mess with/misunderstood the idea of ‘centers’.
I’ve thought that centers, in the z dimension, would be midway through the thickness of the slab. Is it an absolute position, or relative?
Any explanation/suggestions/opinions on this very much appreciated.
Olivera
- (Please ignore the mention of SMD in my files; the name is inherited from my previous attempts to do this using SMD).
run.in (1.83 KB)
PMF-Olivera.colvars (583 Bytes)