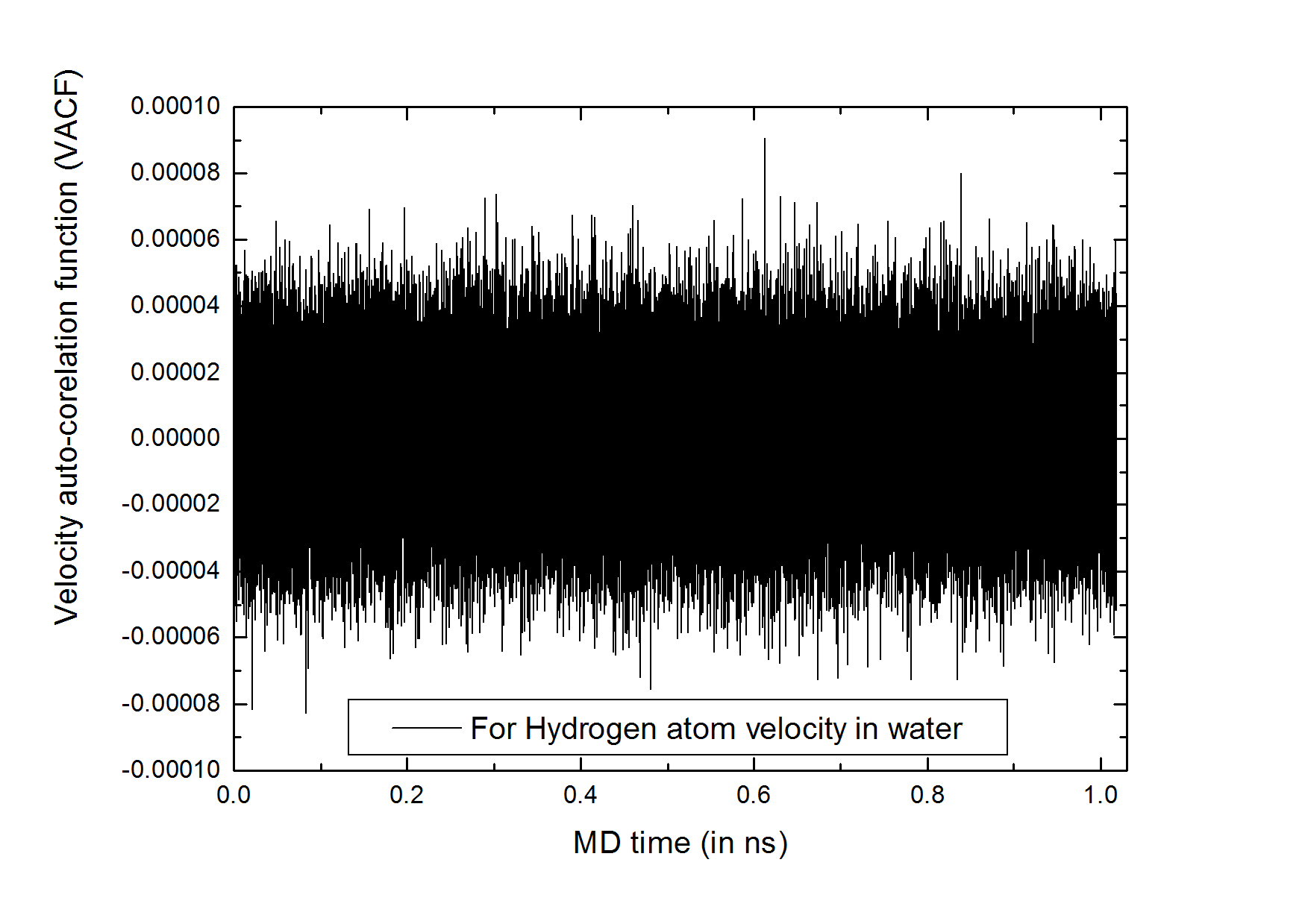
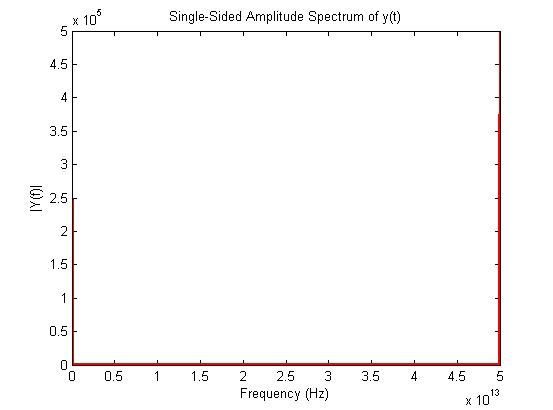
I’m simulating SPC/E flexible water. During the period of equilibration for a timescale of 1 ns, I found that my sample is equilibrated enough, confirmed from different pair-distribution functions and time averaged thermodynamic state variables. But I got problems when I was trying to get the power spectrum of my sample by doing FFT of the VACF of H and O atoms. I just dumped the ACFs of the velocities of H and O atoms in a file and then done FFT in MATLAB, but I got no frequency contains in power spectrum and also the VACF curve looks odd with respect to the published H2O SPC/E flexible ACF curve. Here is my input script for your kind perusal.
units real
dimension 3
boundary p p p
atom_style full
read_data final_water_spc-flexible_data
group ox type 2
group hy type 1
set group ox charge -0.8476
set group hy charge 0.4238
Your setting of ave/time is 1 1 1, meaning the correlation time is 1, so mo wonder you only see the white nosie like vacf.
For flexible water, to resolve the high frequency vibration, time step is suggested not to be larger than 0.25 fs. The correlation time determins the freq grid you are looking at.
Hi Liu,
Thanks for your reply. Yes, I got my mistake in ave/time settings. And I’m using 0.1 fs timestep for safely account the frequency of the flexible water. But how can I calculate and/or guess the correlation time to resolve that high frequency domain ? Is there any rule or idea by which I can make a initial guess ?
the simplest way is to dig out the literature. I did something similar before, so I am sure that someone has done this. Howeve I can not recall the name right now. Spend some time and play with Google scholar.
I’ve tried with ave/time settings “2 1 2” with time step =0.25 fs, i.e the correlation time was 0.50 fs. In this ave/time settings no averaging was done. But in the ACF curve of Hydrogen atom, I saw no convergence to zero. I have attached the curve for your perusal.
BTW I’ve searched and gone through a lot of literature, but have not found one which have given the correlation time for their power spectrum.
Anyway I’m trying with a series from 0.25…0.50…0.75…1.0 fs etc. Any idea ?
Thanks for help. But I also had take this paper as the benchmark but unfortunately they have not given the correlation time for their ACFs curves, they only have written that the time step was 0.2 fs in the last of the computational details para. Anyway thanks again.
I guess that the VACF in your previous email only capture one set. In that case, the curve is of course noisy.
I suggest to match your RDF with the 1998 paper. They did a very concise study on this. Make sure that you have a correct equilibrium state before going to VACF.