I confuse about MP Advanced Correction. If the element is determined, the MP Advanced Correction per atom is a Fixed value? For example, AVO3(A represents all possible elements), no matter which elements the A is, the MP Advanced Correction per atom is 1.628 eV? Or the MP Advanced Correction per atom is always a Fixed value in binary, ternary or multi-nary compounds? If it is, can you send me the list of different elements’ MP Advanced Correction per atom. Thanks! Hope for you reply!
Hi henkekao,
The ‘MP Advanced Correction’ results from the type of simulation techniques we use to generate Materials Project data.
Understanding the ‘MP Advanced Correction’ requires a knowledge of DFT and Hubbard U parameters. The Hubbard U is an additional term applied to DFT calculations to correct for strong on-site Coulomb interactions (elements with highly localized electrons) since GGA DFT does not describe these interactions well.
In the Materials Project, some materials are calculated using a Hubbard U term (these are GGA+U calculations), while other materials do not use a Hubbard U (GGA calculations). The choice of whether to use a U value, and what U value to use, is both element and chemistry dependent.
The ‘MP Advanced Correction’ exists so that energies from GGA+U calculations of one material can be directly compared to energies from GGA calculations of another material. Therefore, the value of the correction depends on both which elements are present, if a Hubbard U term was used for that element in the calculation (and if so, what value of U was used) and the number of atoms of that element present in the simulation cell (typically the number of atoms in the primitive unit cell).
As an example, PbVO3 (mp-25119) has the correction 1.682 eV because of the V atom had a +U term defined and there was a single V atom present in the unit cell whereas YbVO3 (mp-703673) has a correction 6.728 eV (4*1.682) because there are four V atoms present in the unit cell. VCrO3 (mp-770779) has a correction -14.78 eV applied because it has both four V atoms and the four Cr atoms also had a U value defined (each Cr atom has a correction of -2.013 eV applied, so the total for this material is 4*2.013 + 4*1.682).
For any material, you can click the links below “Detailed input parameters and outputs for all calculations” to find more information on the calculations for that material, and this can give information on the what U value (if any) was used. The specific values of the correction for each element are calibrated to the Materials Project and the U values we use, and can be found in the pymatgen code here (link accurate 2018-01-11).
You can find more information on our methods in our publications and in the pymatgen code itself.
Hope this helps!
2 Likes
Thanks for your reply, I think I got your points. It really helps a lot.
1 Like
No problem. Here’s the Materials Project paper going into more detail on the correction and the GGA/GGA+U mixing scheme: https://journals.aps.org/prb/abstract/10.1103/PhysRevB.84.045115 and a pdf available here https://dspace.mit.edu/openaccess-disseminate/1721.1/67053 (see fig 1 and equation 6)
I have read the paper about GGA/GGA+U mixing scheme and I understand how to get the MP Advanced Correction.
First, about the MP Anion Correction, I have a question about Oxidation energies of transition metal oxides within the GGA+U framework: https://journals.aps.org/prb/abstract/10.1103/PhysRevB.73.195107. They recommend a −1.36 eV energy correction for O2 molecule. But in MP, the value is different. By the way, the MP Anion Correction of sulfide is also fitting with the method in the article?
Second, how can we get the MP Gas Correction? I did not find any literature about MP Gas Correction in MP documents.
1 Like
Hi henkekao,
That energy correction comes from constant shift between the calculated and experimental formation energies. In the Wang method, for a combination of practicality and some intrinsic physics, that shift is attributed as a correction to the O2 molecule. Because our +U values are different than that used in the Wang paper, we get a different shift. The sulfide correction is done in the same way. The gas correction is done by fitting the reaction to simple compounds, usually alkali + gaseous compound. We don’t have a paper on it, but this is a good reference: https://journals.aps.org/prb/abstract/10.1103/PhysRevB.87.075150#fulltext
2 Likes
Thinks for your reply again!
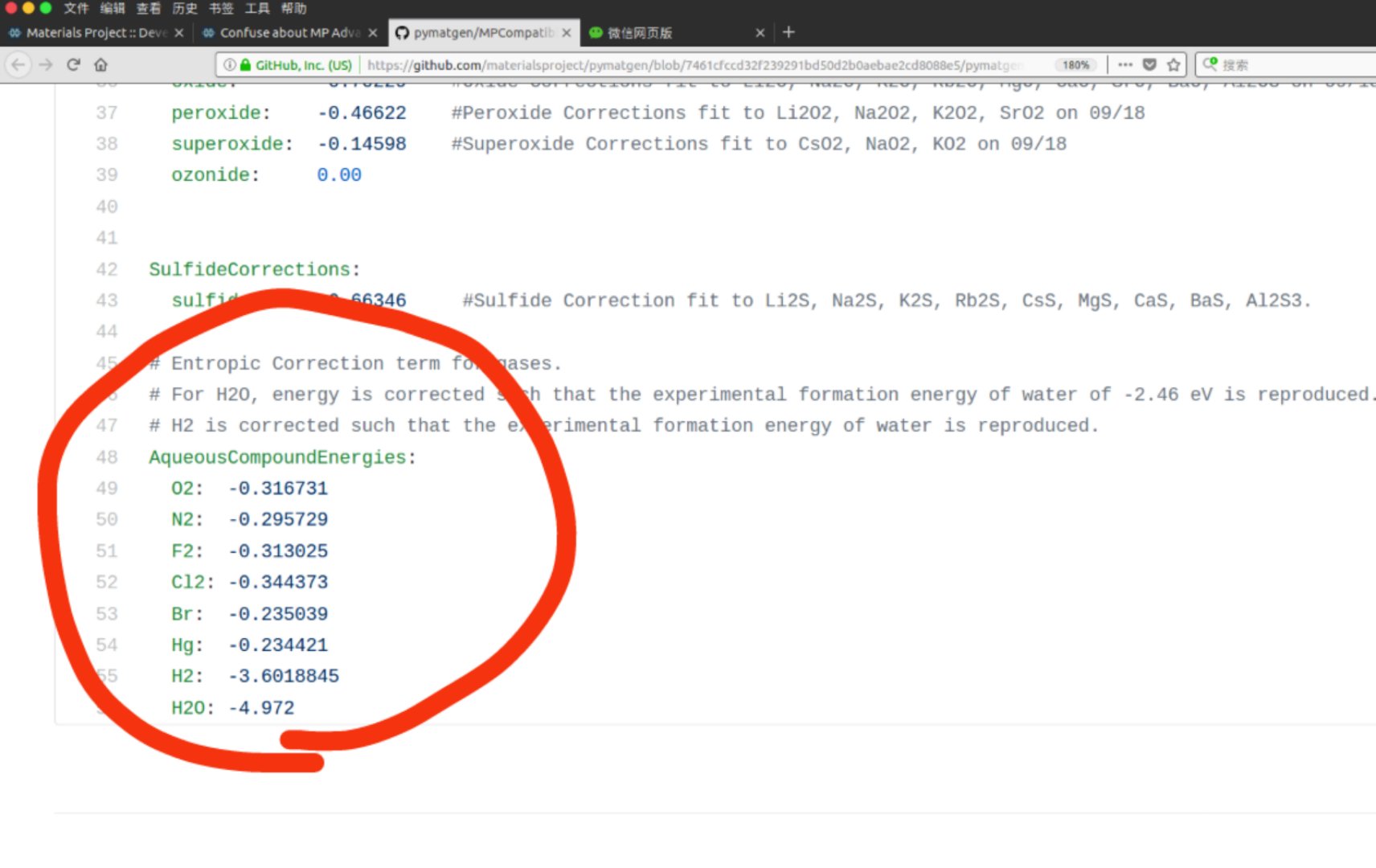
I have read https://journals.aps.org/prb/abstract/10.1103/PhysRevB.87.075150#fulltext, I think this article is still focus on MP Anion Correction instead of MP Gas Correction. So I am still confused about the gas correction. E.g. Cl2, the gas correction in your web is positive 0.5827 eV/per atom, but in your MPCompatibility.yaml is negative 0.344373 eV/per atom. Can you give me a detail of MP Gas Correction. More question, What does ‘AqueousCompoundEnergies’ mean?
Hope for your reply, it is really important for me to know all the details of the total energy and formation energy, because I want to do some research based on your data in MP!

Hi Henkekao,
The corrections in MP try to recreate the same stability in our data set as measured experimentally. We do this by trying to get reaction energies correct, since these should be temperature agnostic to first approximation. The Anion and Gas corrections are found in basically the same way. You take a set of well measured reactions and compare with what MP predicts based on uncorrected values and account for the difference. Which correction you get depends on which reactions you analyze. It’s also really important not to double count.
The MP Gas correction changes the energy of gaseous compounds to force them to have a certain formation energy. The section you want to look at is CompoundEnergies, which has the desired gaseous formation energy for various compounds including Cl2. It doesn’t include O2, because we correct for this by modifying the compounds, such as Al2O3, rather than the gaseous element itself due extraneous binding of the O2 molecule in DFT.
So far, we’ve talked about global stability not really accounting for the environment. If we want to talk about stability in water, where gas is dissolved, then some of those corrections have to change. The formation energies for dissolved gas in water is different than gas just floating about and the AqueousCompoundEnergies are those formation energies. This is used in our Pourbaix to plot stable phases as a function of pH and Voltage.
1 Like