Thank you very much Axel sir for helping me again to resolve my first
problem.
But I am still facing the second problem which I mentioned earlier and it
do not seems to be a Ovito problem. I am using "Color Coding" function of
Ovito to assign color on the basis of mass of the atoms.
if you see it only in ovito, it is an ovito problem.
to make people have a look into whether there is a problem in LAMMPS you
have to provide more convincing proof.
- you have to demonstrate that the dump files already contain incorrect
mass information
- you have to demonstrate this unexpected behavior can be reproduced with
the very latest LAMMPS version (19May2017 at the time of this writing)
until then, we have to assume, that the problem is not in LAMMPS.
here is my proof. with this minimal input:
region box block 0 1 0 1 0 1
create_box 2 box
create_atoms 1 single 0.5 0.5 0.0
create_atoms 2 single 0.0 0.5 0.5
mass 1 4.0
mass 2 2.0
write_dump all cfg two.cfg mass type xs ys zs
i get exactly the masses, that i have assigned in the .cfg file:
Number of particles = 2
A = 1 Angstrom (basic length-scale)
H0(1,1) = 1 A
H0(1,2) = 0 A
H0(1,3) = 0 A
H0(2,1) = 0 A
H0(2,2) = 1 A
H0(2,3) = 0 A
H0(3,1) = 0 A
H0(3,2) = 0 A
H0(3,3) = 1 A
.NO_VELOCITY.
entry_count = 3
4.000000
C
0.5 0.5 0
2.000000
C
0 0.5 0.5
i see no indication that LAMMPS is mixing things up and i would be
extremely surprised of this, considering how many people use the cfg file
format and atomeye to visualize LAMMPS output. if LAMMPS would be creating
incorrect output, it would have long been noticed and reported, since the
code for dumping .cfg style files has been unchanged for quite a while.
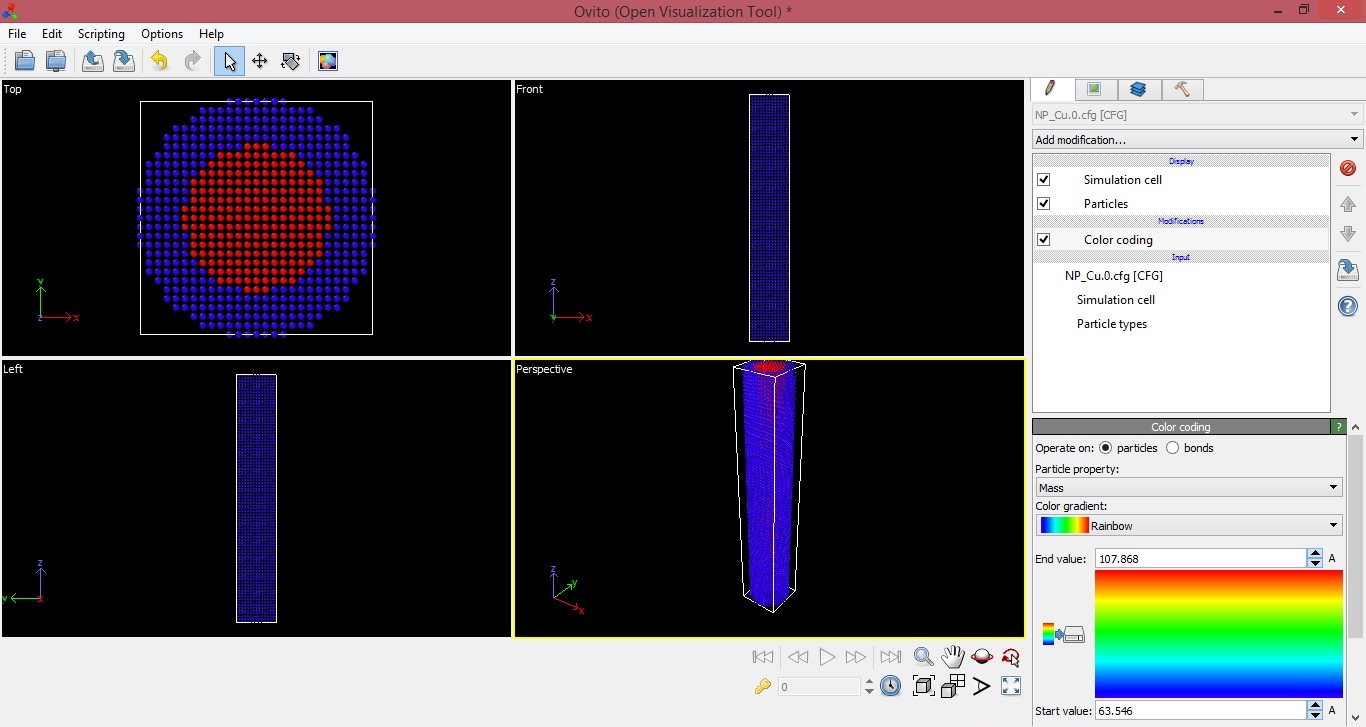
According to that, it is clearly showing from the image (screenshot
attached) that my Ag has formed the core and Cu has formed the shell
(instead of Cu core and Ag shell). The same information I obtained by
putting my cursor on atoms of respective region and it also shows that the
mass of the particles of core is of Ag and mass of the particle of shell is
of copper.
please note the following comment from the dump documentation about the cfg
format output:
Note that you will typically want to use the *dump_modify element*
<http://lammps.sandia.gov/doc/dump_modify.html> command with CFG-formatted
files, to associate element names with atom types, so that AtomEye can
render atoms appropriately.
axel.