Hello all:
I am doing NPT simulation (fix npt ) of Polymer in electrolytes.
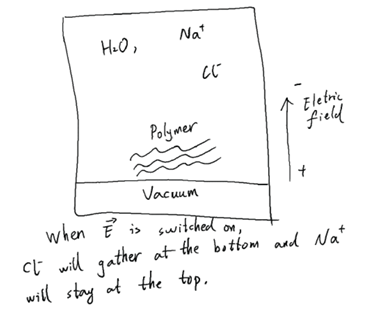
I want to create a vacuum at the bottom of the simulation box and all atoms cannot go into. The situation is shown in the figure. What is the best way of doing this? If I do not use a vacuum, and use a “fix wall” and “boundary p p f” to build a wall at the two faces of Z-axis, could the Cl- ions at the bottom still interact with the Na+ ions at the top?
I want to use a vacuum because if I apply electric field along Z-axis, ions will be driven to the top or bottom of the box and they will not cross the boundary. The polymer is at the bottom of the box by using “fix spring”.
By the way, if there is no vacuum and when electric field is used along Z-axis, will ions gather into the bottom and top of the box?
I mean, although there is an electric field pushing positive ions (Na+) up and negative ions (Cl-) down. But because of periodic boundary conditions, the Na+ ions and Cl- ions are getting closer and will eventually cross the Z boundary. Is this interpretation correct or wrong?
As you have already discovered, using fix efield with periodic boundary conditions can lead to inconsistent and problematic behavior. So if it is possible to set up a fixed (or shrinkwrap) boundary, you should do that. However, since you have ions, you probably also want to use a kspace solver, and then you have a problem, since despite entering a “p p f” boundary, internally you will still have “p p p” plus a significant chunk of vacuum.
This latter property is also important to keep in mind for the answer to your question of whether charges across the z-boundaries will “see” each other. With a plain cutoff, they will not. With kspace and thus long-range coulomb, you have to request to use the slab correction which will do two things: 1) it will expand your box in z-direction by a factor of 3 (or whatever else you specify). This added box volume must remain empty; no atoms may ever enter, so either a wall fix or fix oneway are required. 2) the net dipole of your slab in z direction is computed and then an Poisson solver applied to compute the interaction of this dipole with its periodic images and this is then subtracted from the kspace forces so that all charged particles do not “see” the effect of the periodic images in z direction. This is a bit of an approximation, since it only considers the dipole and only the z-component, but systems are usually large enough that other components or quadrupole or higher order interactions can be neglected.
Regardless of the presence of a vacuum. The impact of fix efield is effectively that of an instance of fix addforce, where the force magnitude is scaled by the (partial) charge of the individual atom.
Yes I am using kspace, so I need to use “boundary p p p”
I think of some possibilities and questions:
I can try “fix wall” but with periodic boundary, so ions cannot across the boundary and I can still use kspace.
I can try “fix oneway”
can I use “region” command, and “create_atom” in the defined region so that it works as a slab to prevent ions crossing the boundary? When running the NPT, i just “fix npt” for the polymer+electrolyte, but not the Slab.
I have try this way, but the code some don’t run with GPU.
No, as already explained. You can use boundary p p f but then also must use kspace_modify slab 3.0 or similar. In that case you should not use fix npt. There is no reason for it.
You must have sufficient vacuum between the periodic images and apply the slab correction to the kspace solver or else you will have unwanted interactions.
In this setup, you use a pair of positively and negatively charged walls to create an electric field across your cell – you then repeat the entire system in reverse to create a dipole-neutral supercell.
Hi Srtee:
Thank you for the idea. I want the electric field to be the same every in the box. It seems that add some charges on the wall cannot create such an electric field. Does Lammps have commands to do that?
it will expand your box in z-direction by a factor of 3 (or whatever else you specify). This added box volume must remain empty; no atoms may ever enter, so either a wall fix or fix oneway are required.
Hello Axel:
When I try to use
boundary p p f
kspace_modify slab 3.0
I get the error “ERROR on proc 2: Angle atoms 105978 105980 105979 missing on proc 2 at step 16 (src/ntopo_angle_all.cpp:64)”. What is the reason?
Also, there is a note in the documentation that " If you wish to apply an electric field in the Z-direction, in conjunction with the slab keyword, you should do it by adding explicit charged particles to the +/- Z surfaces. If you do it via the fix efield command, it will not give the correct dielectric constant due to the Yeh/Berkowitz (Yeh) correction not being compatible with how fix efield works."
How to add explicit charges to the Z-surface? the only charges is Na+ and Cl- in my system?
I want to use a vacuum because if I apply electric field along Z-axis, ions will be driven to the top or bottom of the box and they will not cross the boundary. The polymer is at the bottom of the box by using “fix spring ”.
Hello again:
I think of a way: I want to use change_box to enlarge the Z boundary at the top and bottom of the box, then use region to create a region, then use fix oneway to force atoms not entering the region.
Does the scheme work for the picture described in the 1st post?
That kind of error is a common error (search the mailing list archive or just google the message with “lammps” added) and can have multiple reasons. It is an indication of bad simulation dynamics most often caused by an unsuitable initial configuration when this happens after a few steps, or unsuitable force field parameters when it happens later, or a combination of both. It can also happen, if your choice of fix efield parameters is unphysically large.
You would do what @srtee has suggested (but with a difference geometry) and add another type of atom to the system that is not included in time integration and put them (with opposite charges) at both ends of the simulation cell. Check out the referenced publication.
For as long as you use a kspace solver you will have interactions across periodic boundaries. Since the Coulomb potential decays with 1/r, you would have to add a very large amount of vacuum to make those interactions decay to a negligible level. The computational cost for a kspace solver, however, mostly scales with the volume of the cell, and less so with the number of atoms and thus this is a costly approach. The approach suggested by @srtee tries to counter that.
I could be cheeky and tell you to build a pair of flat conducting graphene electrodes using the plugin that I maintain (GitHub - srtee/lammps-USER-CONP2: updated constant potential plugin for LAMMPS). That will generate two equipotential surfaces which will ensure a constant electric field across the cell (and a nice citation for me).
But I suspect you do not need the electric field to be that uniform, and you do not need the extra computational cost required, and therefore a few grids of constant charge particles will do the trick. You need to work out what level of detail is acceptable. After all, there are many other approximations you are already making, such as the water model, the ion models, the polymer model, and the thermostat.
If you read the reference I gave, you will see that they mention “the electric field lines are not exactly perpendicular near the charged particles used to generate the field”. And in that paper they are okay with that, because they are mainly interested in studying the behavior of a liquid-liquid interface far from those charged particles. You need to work out what exactly you are interested in and how accurate your electric field needs to be to observe that, and then it will be much clearer to you what to do.
Regarding your “atoms missing” error – if your system ran fine with “boundary p p p” and is now losing atoms under “boundary p p f”, one likely possibility is that you have atoms going across the fixed boundaries and therefore getting lost.
Eventually I achieved my purpose of gathering Na+ at the bottom and Cl- at the top without crossing the boundary, by inserting a Si slab at the box bottom, then I fix NVT for the system excluding Si (Si atoms are not integrated and stationary in MD). As the Si slab is large enough, the Coulomb force of Cl- at the other side can be neglected for the problem I am studying.
Some remark: for the fix oneway method without fixing a wall, water molecules have to be wrapped to avoid broken bonds across the periodic boundary. Srtee’s @srtee method is worth trying out for simulation enthusiasts but obviously more complicated than the Si slab.