Dear lammps developers,
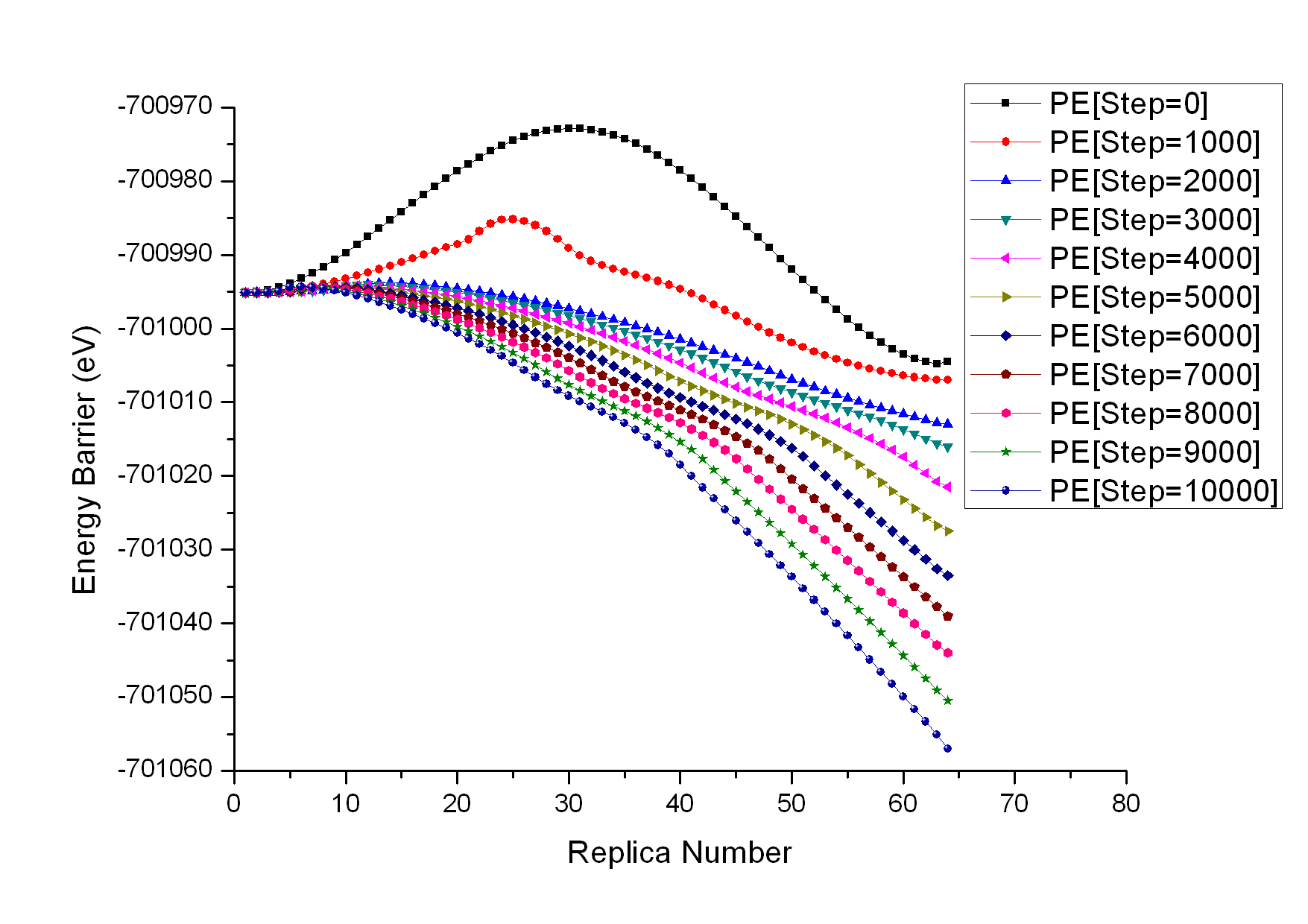
I am calculating the energy barrier of dislocation nucleation in metal nanowires. I have attached one of my results (graph 1).
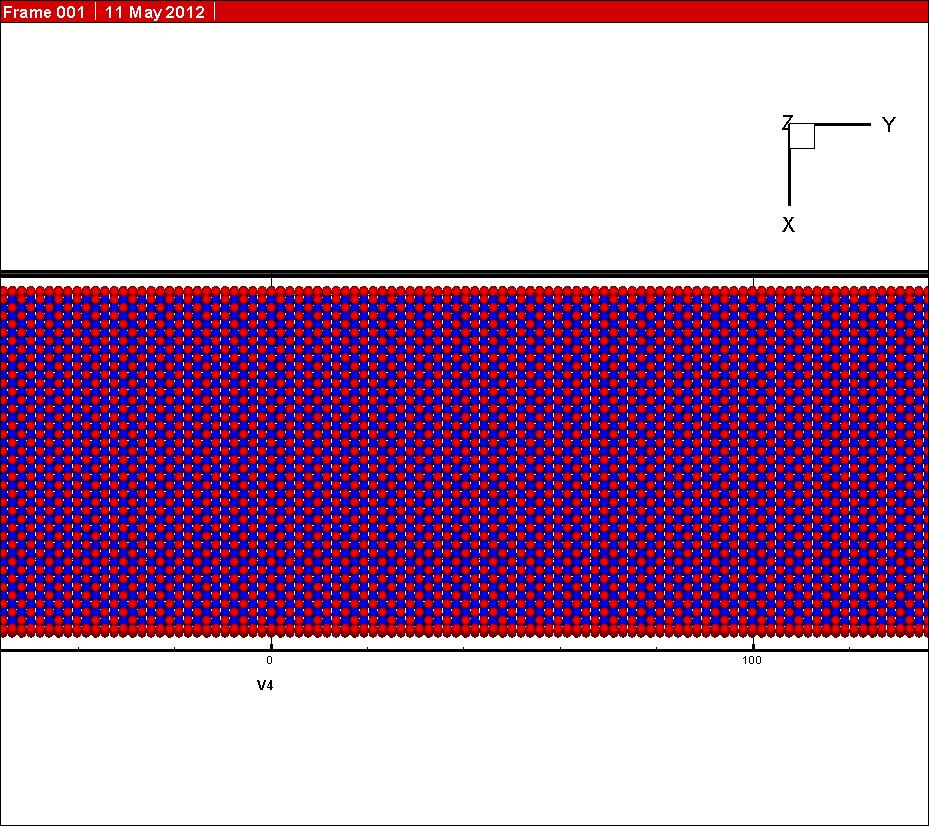
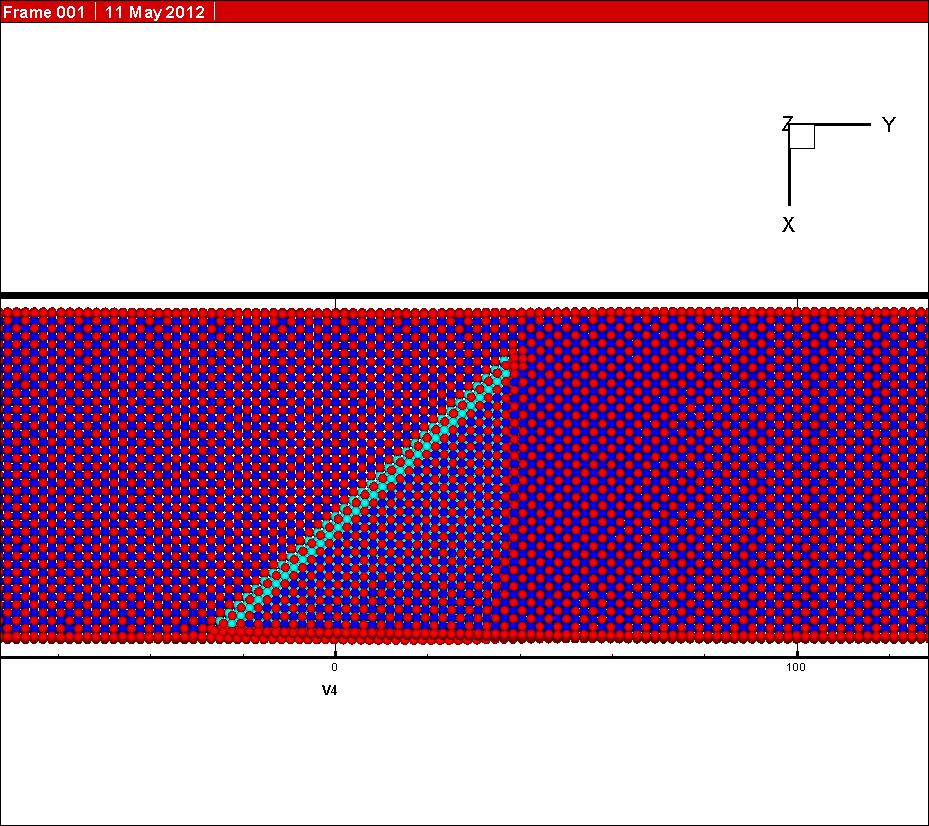
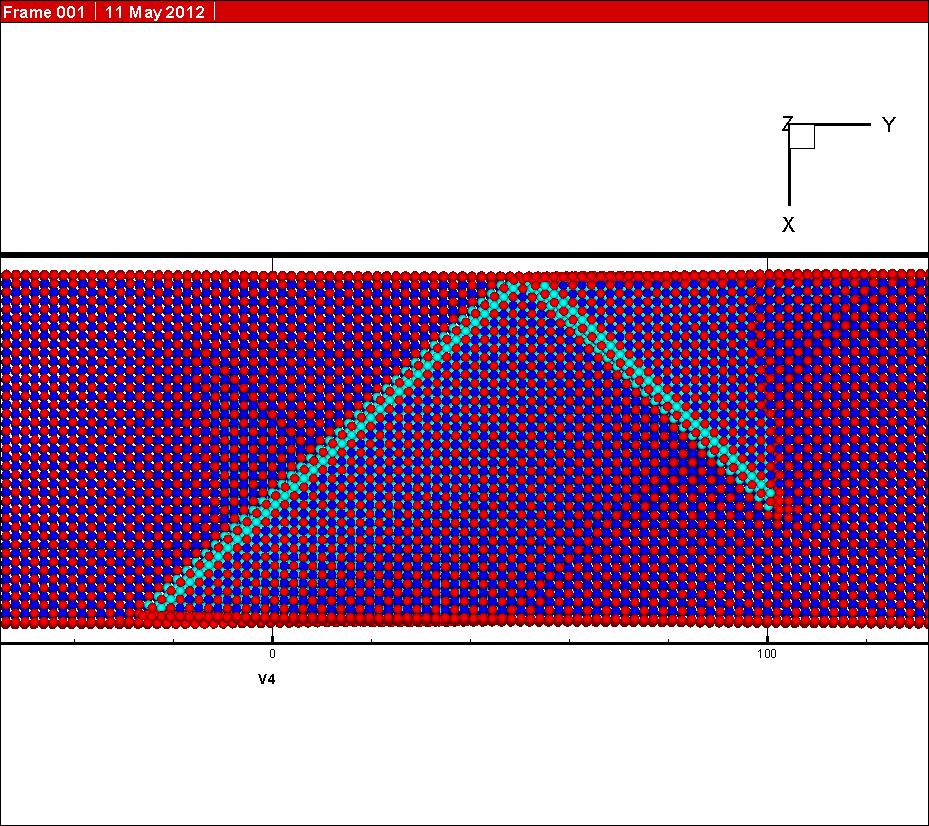
In my simulation, the initial state (see graph 2 attached) and the final state (see graph 3 attached) have both been relaxed before NEB calculation. However, one can see from the graph attached that the final state have changed during simulation (see graph 4 attached). I know the final state was driven to find the minimum energy state during the NEB calculation, for it is not the minimum energy state at the beginning of my simulation.
However in my simulation, I just want to find the energy barrier between the initial and the final states, and it will be better for the two states to be fixed. Meanwhile, I realize in free end NEB calculation, the final state is fixed. Therefore, I want to modify the neb.cpp or the fix_neb.cpp in REPLICA package to fix the initial and the final state. Is it easy for me the do such modify? Could you give me some suggestions about which code should be modified?
Thank you.
H. Wei