I´ve been trying to fit some cross Lennard-Jones parameters between a molecule and a rigid framework using GULP.
I successfully fitted a potential with reasonable accuracy, but I´ve noticed that some forces are zero within the force field while not zero in my observables (DFT calculations).
Here is a picture to illustrate:
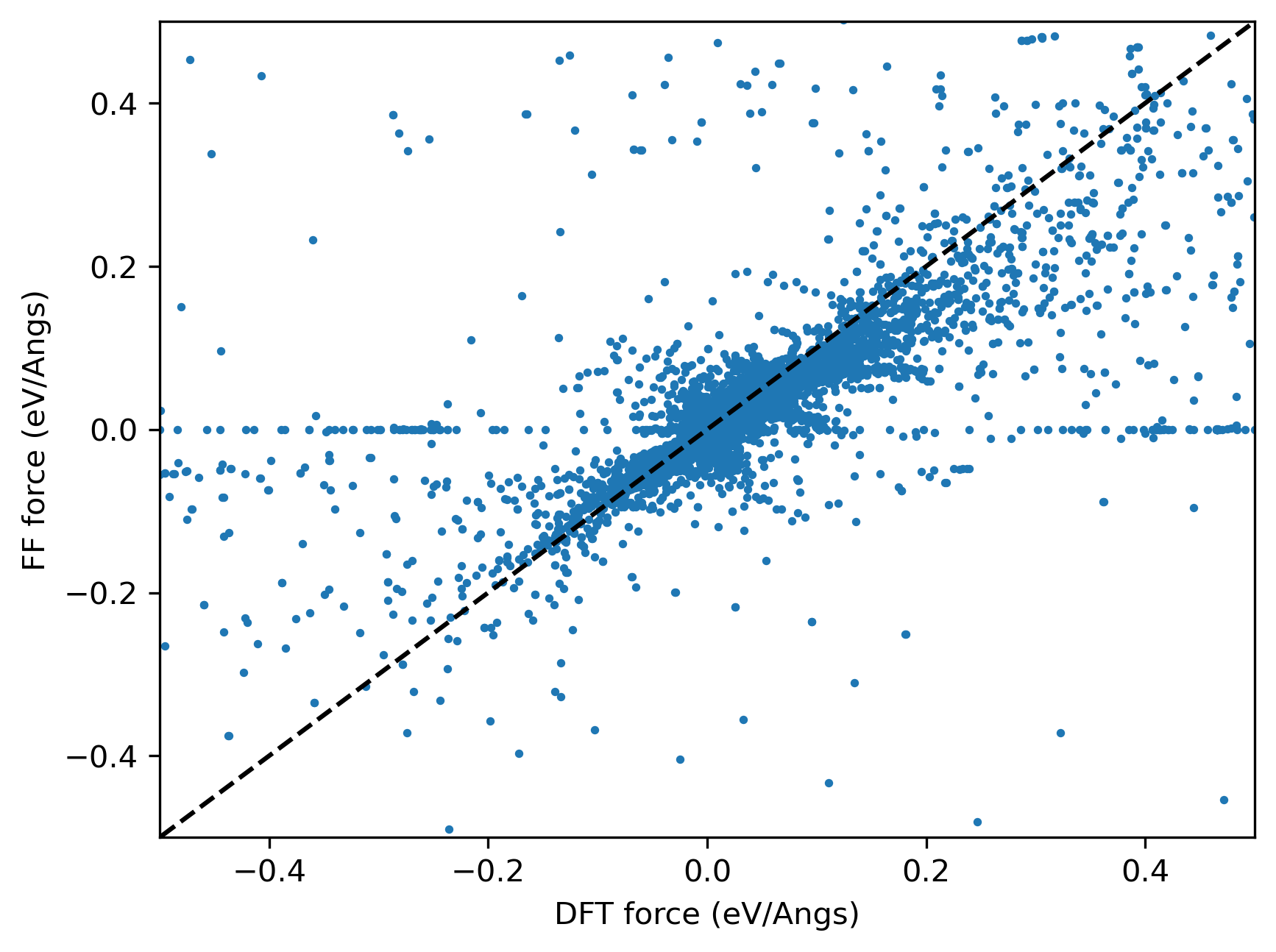
In the process of trying to understand this behavior, I have noticed that for some reason the first line of the gradients section is not considered in the fitting.
You can see this by looking at the output when the observables are printed:
81 Energy -0.403059 349.1410 81
82 Energy -0.289607 4.3360 82
83 Derivative 0.001900 0.0000 2 x 1
84 Derivative 0.001950 0.0000 2 y 1
85 Derivative -0.001238 0.0000 2 z 1
I have double-checked by printing in the setcfg.F90 file the lines where the fgrad is assigned to fobs.
Is there a particular reason for this?
Thank you in advance!