Hello,
I was trying to calculate the self diffusion coefficient for the pure metals like Molybdenum, Tungsten or Tantalum.
I am using the following program to obtain MSD and RDF at different temperatures.
units metal
dimension 3
boundary p p p
atom_style atomic
variable latparam equal 3.1652
----------------------- ATOM DEFINITION ----------------------------
lattice bcc {latparam}
region whole block 0 31.5 0 31.5 0 31.5
create_box 1 whole
lattice bcc {latparam} orient x 1 0 0 orient y 0 1 0 orient z 0 0 1
create_atoms 1 region whole
pair_style eam/alloy
pair_coeff * * MoTaWTiZr.set W
min_style cg
min_modify dmax 0.1
thermo 10000
thermo_style multi
minimize 1e-15 1e-15 1000000 1000000
compute csym all centro/atom bcc
compute peratom all pe/atom
reset_timestep 0
timestep 0.001
velocity all create 4000 123456 dist gaussian
fix 1 all npt temp 4000 4000 1 iso 0 0 1 drag 1
thermo 1000
thermo_style custom step temp press ke pe etotal vol density
run 90000
compute myRDFall_1 all rdf 50
fix 116 all ave/time 100 1 100 c_myRDFall_1[*] file tmp1_all.rdf mode vector
compute msd1_all all msd com no
compute vacf1_all all vacf
fix 5 all vector 1 c_vacf1_all[4]
variable vacf_all equal dt*trap(f_5)
thermo_style custom step temp v_vacf_all c_msd1_all[4]
thermo 5
run 5000
unfix 1
fix 10 all npt temp 4000 3300 1 iso 0 0 1 drag 1
thermo 10000
thermo_style custom step temp press ke pe etotal vol density
run 2000000
unfix 10
fix 11 all npt temp 3300 3300 1 iso 0 0 1 drag 1
thermo 1000
thermo_style custom step temp press ke pe etotal vol density
run 50000
compute myRDFall_2 all rdf 50
fix 216 all ave/time 100 1 100 c_myRDFall_2[*] file tmp2_all.rdf mode vector
compute msd2_all all msd com no
compute vacf2_all all vacf
fix 9 all vector 1 c_vacf2_all[4]
variable vacf2_all equal dt*trap(f_9)
thermo_style custom step temp v_vacf2_all c_msd2_all[4]
thermo 5
run 1000
unfix 11
fix 18 all npt temp 3300 2883.8 1 iso 0 0 1 drag 1
thermo 10000
thermo_style custom step temp press ke pe etotal vol density
run 2000000
unfix 18
fix 19 all npt temp 2883.8 2883.8 1 iso 0 0 1 drag 1
thermo 1000
thermo_style custom step temp press ke pe etotal vol density
run 50000
compute myRDFall_3 all rdf 50
fix 316 all ave/time 100 1 100 c_myRDFall_3[*] file tmp3_all.rdf mode vector
compute msd3_all all msd com no
compute vacf3_all all vacf
fix 9 all vector 1 c_vacf3_all[4]
variable vacf_all equal dt*trap(f_9)
thermo_style custom step temp v_vacf_all c_msd3_all[4]
thermo 5
run 1000
unfix 19
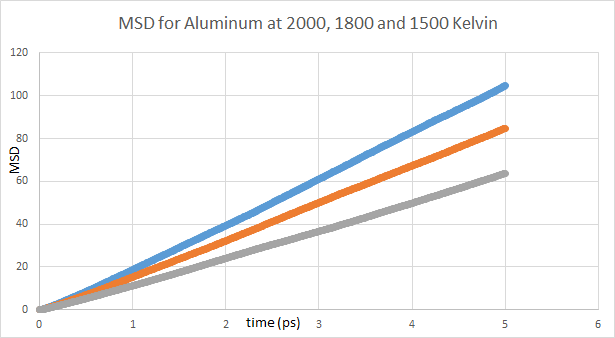
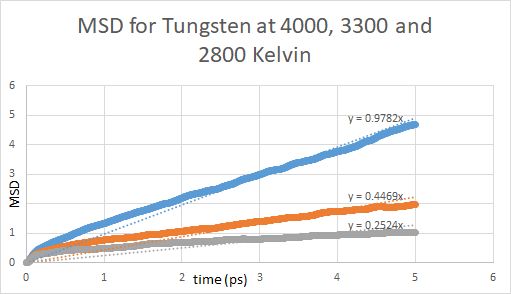
The problem that I am facing is that the MSD values are extremely small (~1 sq Angs/atom in 1 ps) as compared to MSD values in most publications (~100 aq Angs/atom). This is making the D_0 (temperature independent pre-exponential for diffusion) value 2-3 orders of magnitude smaller than the values mentioned in literature.
I agree this is not a program related issue but I would like to hear from your vast experience, any advice that I should follow for getting better results of MSD. I am attaching both the input file and log.lammps files for your reference.
input_W.txt (5.52 KB)
log.lammps (379 KB)