Hello,
Is it possible to use displace_atoms rotate on molecules which partially cross periodic boundaries?
It works well when I don't have anything crossing periodic boundaries, but when I use displace_atoms rotate the parts of the molecules which crossed periodic boundaries are not rotated correctly, although the rest of the molecule is.
I thought it might work if I rotated based on xu, yu, zu instead but I don't seem to have access through a variable. I think I basically want to unwrap, then displace_atom rotate then it can be wrapped back, although LAMMPS probably already does this step.
Thanks.
Josh
Hello,
Is it possible to use displace_atoms rotate on molecules which partially
cross periodic boundaries?
It works well when I don't have anything crossing periodic boundaries, but
when I use displace_atoms rotate the parts of the molecules which crossed
periodic boundaries are not rotated correctly, although the rest of the
molecule is.
this sounds like a bug. the documentation for the displace_atoms command
states that arbitrary displacements over long distances should be possible.
if you are not using the very latest patchlevel of LAMMPS, please double
check, that this unexpected behavior still exists.
if confirmed, can you then please construct a minimal example input and
upload the input deck with a short description to
https://github.com/lammps/lammps/issues and open a new issue.
thanks,
axel.
Thanks Axel,
I added the issue to github with the example. I tested with the latest
github unstable branch. I see the same issue as the Feb 21 17 version I
was using.
thanks. much appreciated.
i've been expecting that this persists, but it is very helpful to have this
confirmation.
hopefully somebody has the time to look into this soon.
axel.
Hello,
I think I understand what the issue is.
The rotation wasn’t based on the unwrapped coordinate. So I changed
Lines 277-279 from
d[0] = x[i][0] - point[0]
d[1] = y[i][1] - point[1]
d[2] = z[i][2] - point[2]
to
domain->unmap(x[i],image[i],unwrap);
d[0] = unwrap[0] - point[0]
d[1] = unwrap[1] - point[1]
d[2] = unwrap[2] - point[2]
with the appropriate variable definitions.
Later these displacements are used to redefine x[i][], so the new x[i][] are all unwrapped but now have the wrong image flags.
This appears to cause all of the particles to rotate correctly, but since I didn’t change the image flags, the image flags are wrong and once the remapping is done on line 333, it appears to add to the image flags instead of resetting them from scratch.
Is there an easy way to reset all the image flags, since after the unwrapping above, all the image flags should be 0, and then should correctly be assigned if they were wrapped?
Would I need to do something like what read_dump does when resetting image flags?
Thanks.
Josh
since the code is overwriting the coordinates later, there is no need to
store the unwrapped coordinates in a separate array.
resetting the image flags should also be done at the same time. thus i
would propose the following change:
diff --git a/src/displace_atoms.cpp b/src/displace_atoms.cpp
index 46f697f..7db7e21 100644
--- a/src/displace_atoms.cpp
+++ b/src/displace_atoms.cpp
@@ -271,9 +271,15 @@ void DisplaceAtoms::command(int narg, char **arg)
int *body = atom->body;
int *mask = atom->mask;
int nlocal = atom->nlocal;
+ imageint *image = atom->image;
for (i = 0; i < nlocal; i++) {
if (mask[i] & groupbit) {
+ // unwrap coordinate and reset image flags accordingly
+ domain->unmap(x[i],image[i]);
+ image[i] = ((imageint) IMGMAX << IMG2BITS) |
+ ((imageint) IMGMAX << IMGBITS) | IMGMAX;
quick update: the proposed patch seems to be working as expected and is now included in pull request #495
https://github.com/lammps/lammps/pull/495
axel.
Hi Axel,
Thank you. Resetting the image flags was the part I wasn’t sure how to do.
It seems like this should work, but I’m still seeing what I think are the wrong image flags for the example I posted. See attached screenshots where step1 after the rotation shows a huge bond length. Were you getting it to work?
Thanks.
Josh
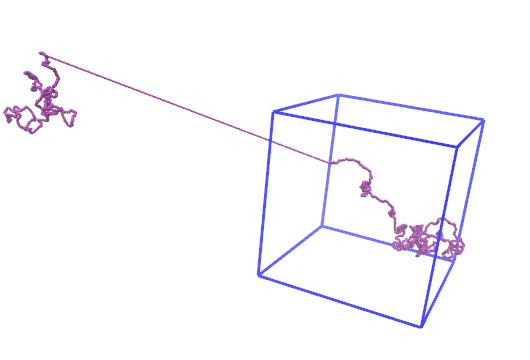
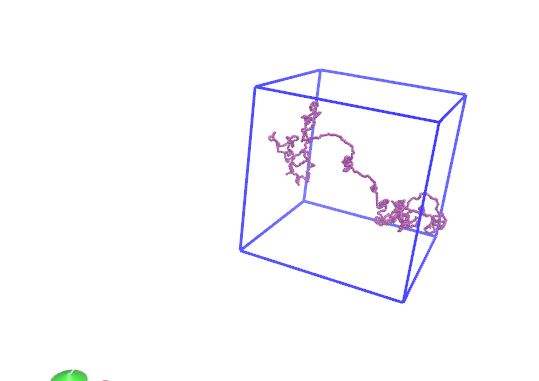
Hi Axel,
Thank you. Resetting the image flags was the part I wasn't sure how to do.
It seems like this should work, but I'm still seeing what I think are the
wrong image flags for the example I posted. See attached screenshots where
step1 after the rotation shows a huge bond length. Were you getting it to
work?
the molecule in the example you posted, is mostly outside the box in the
z coordinate. the selected position to rotate about, however, is a wrapped
atom coordinate. the rotation you see is correct, albeit unexpected. if you
move your molecule inside the box, with the following command:
displace_atoms all move
0.0 0.0
210.4261081
you get the expected transformation.
axel.
correction. there is a typo. the transformation must be:
displace_atoms all move
0.0 0.0
-210.4261081
alternately, you can “unwrap” the point to rotate about by adding 210.4261081 to its z-coordinate:
variable zi equal z[v_i]+210.4261081
in both cases, you should get the expected rotation around that location.
if you want a more generic solution, you need to generate a group with the atom to rotate about and can obtain it unwrapped position through the xcm() function. e.g. like this:
Random point of axis of rotation (centered on random atom i)
group rcenter id ${i}
variable xi equal xcm(rcenter,x)
variable yi equal xcm(rcenter,y)
variable zi equal xcm(rcenter,z)
axel
Works perfectly!
Thank you very much.
I totally spaced that I was rotating a center on a wrapped atom, not my intention. Thank you for pointing that out.
Works perfectly!
Thank you very much.
I totally spaced that I was rotating a center on a wrapped atom, not my
intention. Thank you for pointing that out.
that is easy enough to overlook. i often spot these kind of issues,
because the same thing has happened to me before. 
axel.