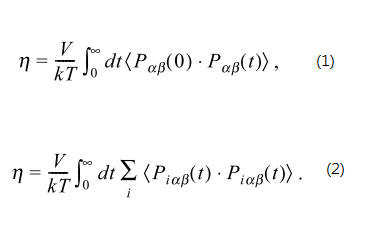Dear LAMMPS developers and users,
In the sample script of calculating viscosity via Green-Kubo formula, the pressure tensor is calculated as a system value:
http://lammps.sandia.gov/doc/Section_howto.html#howto_21
Yet, I would like to try another approach, namely, treat the pressure as per-atom value, do the auto-correlation, sum them up, and then integrate by time.
That being said, do equation 2 instead of equation 1 in the attached image file.
compute stress/atom gives the instantaneous pressure of each atom, but does fix ave/correlate treat arrays rather than single values?
Or is there any better way to perform this calculation?
Thank you for your attention. Any comment/suggestion is greatly appreciated.
LC Liu
ps. below is the reference talking about these two types of Green-Kubo formula.