Dear lammps users,
Sorry to send a wrong reply email before. I have questions about the energy drift of equilibrating system.
I ran a simulation and the equilibrated at different time-step. However, the energy results of same system show the different energy at different time-step. Then I ran a example script in the lammps example, the energy results of the example system has the similar problem.
I know the “shadow” Hamiltonian may cause various energy results of same system at different time-step. Did the lammps compute the actual Hamiltonian mechanics of the system? Can anybody give any comments?
Kerwin
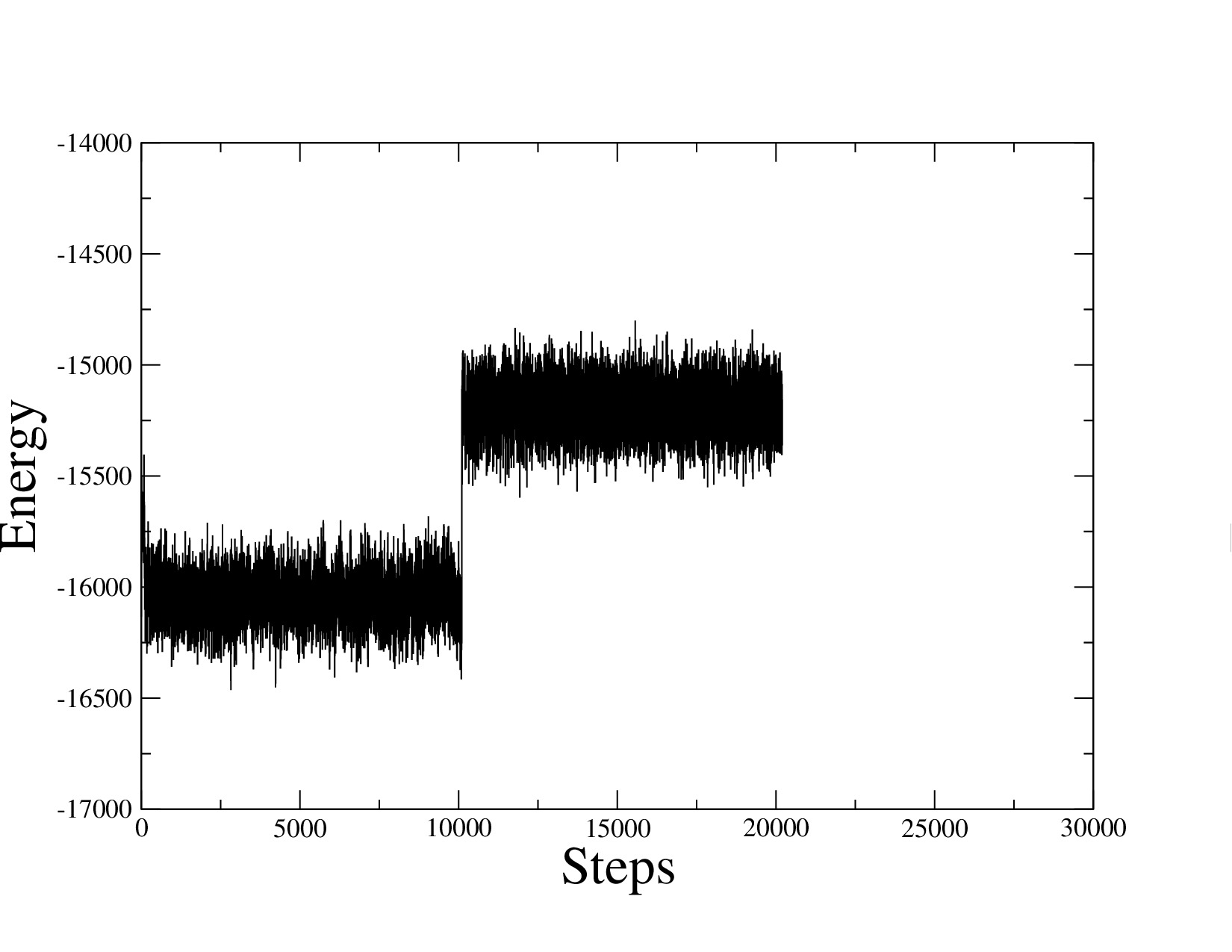
This most likely is not drift, because it looks like a sudden jump at a nicely round number of steps (10000). Which example did you run? Are all your pair styles and settings restart-compatible? If you do not restart but run twice as long, is the jump present or not?
I ran a example in.deca-ala_imd under directory lammps-stable_11Aug2017/examples/USER/misc/imd.
I didn’t change anything of example script except inserting restart command. I changed the time-step of system at 11000 step, then the energy jumped. I also checked the energy of the system, and is was continued in the whole simulation. Once I increase time-step, the energy increase.
I just ran the simulation in one file without restart command. The energy of the system also increased when time-step increase.
This most likely is not drift, because it looks like a sudden jump at a nicely round number of steps (10000). Which example did you run? Are all your pair styles and settings restart-compatible? If you do not restart but run twice as long, is the jump present or not?
This sounds like it might be specific to interactive MD simulations then, but I am not sure. Axel is more of an expert on that I think.
It just seems to me that it is weird that your energy should jump such a specific value upon a time step change.
Energy drift, however, is characterized by a very slow and gradual change of the total energy of the system, which is not at all what you are seeing.
This sounds like it might be specific to interactive MD simulations then,
but I am not sure. Axel is more of an expert on that I think.
but i cannot say anything, because the description being given is
ambiguous. it is not clear what modifications were made to the example
input, and whether the restarting was done correctly. in the vast majority
of these cases, there is a mistake or misunderstanding in there somewhere,
but without a crystal ball, it is impossible to detect and to give advice.
axel.
Thanks for the current comment.
I did the test as Stefan said, ran the simulation at different time-step without restart command. For this test simulation, I remained all the parameter and setting ran short steps, but didn’t allowed the fix shake and fix imd commands. The simulation was only ran in npt ensemble at different time-step. And the figure clear show the energy jumped when time-step changed.
The modified input script is posted here. And the new energy result figure was attached. Can you point out if there is anything wrong?
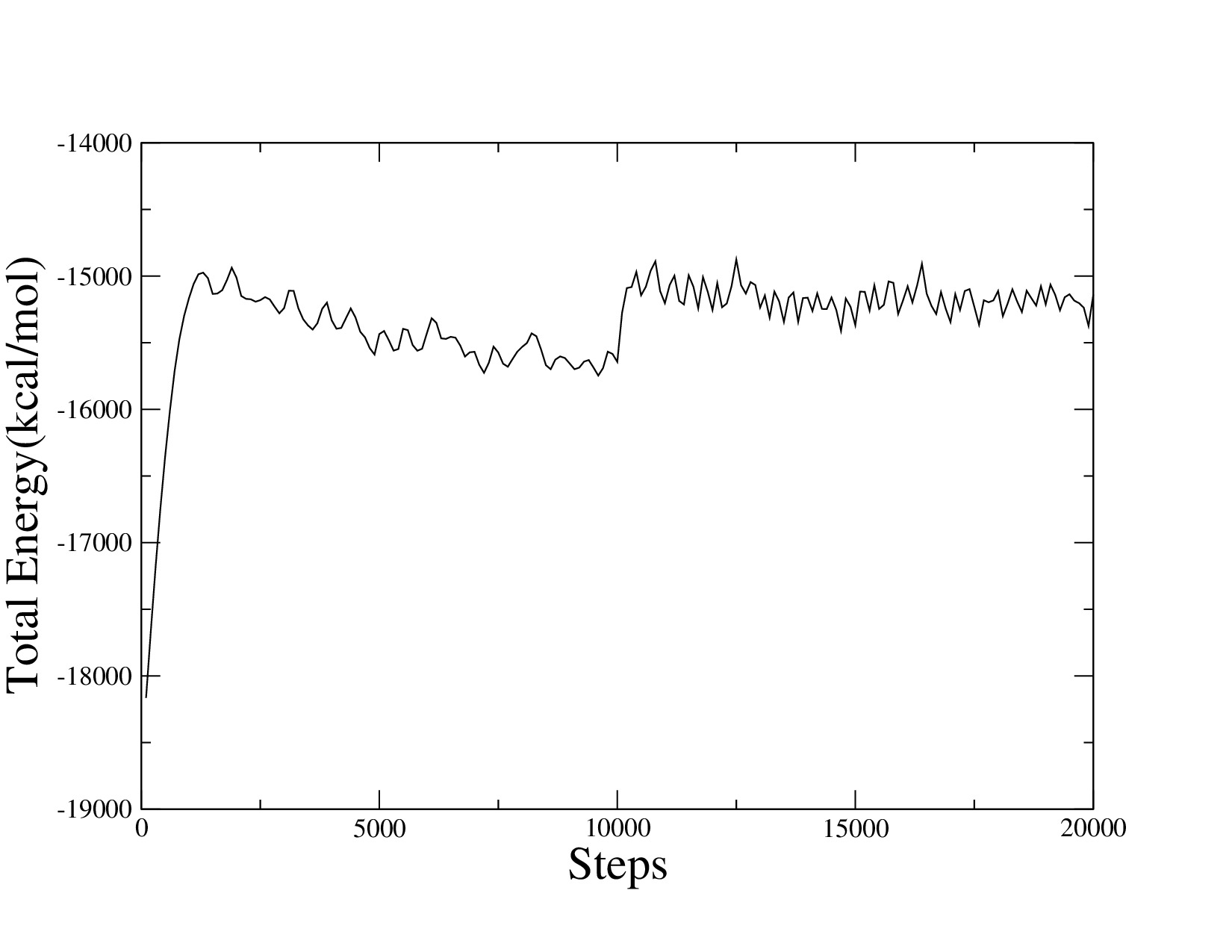
Thanks for the current comment.
I did the test as Stefan said, ran the simulation at different time-step
without restart command. For this test simulation, I remained all the
parameter and setting ran short steps, but didn't allowed the fix shake
and fix imd commands. The simulation was only ran in npt ensemble at
different time-step. And the figure clear show the energy jumped when
time-step changed.
The modified input script is posted here. And the new energy result figure
was attached. Can you point out if there is anything wrong?
you've commented out fix shake. even a time step of 1fs is too large for
that scenario. but cranking up the time step to 2fs explains the step in
(average) energy.
axel.