Hello everyone,
I am working on simulation of a polymer system at high temperature using ReaxFF. When I subject my polymer (~9000 atoms confined in a box) to high temperature, it exploded near 935K. I am trying to figure out a reason for that.
While the other ReaxFF energies (bond energy, angle energy, lone pair energy etc.) did not change significantly during explosion, one parameter (charge equilibration energy, Eqeq) did change significantly. Here’s a graph of final 10 data points of Eqeq before explosion:
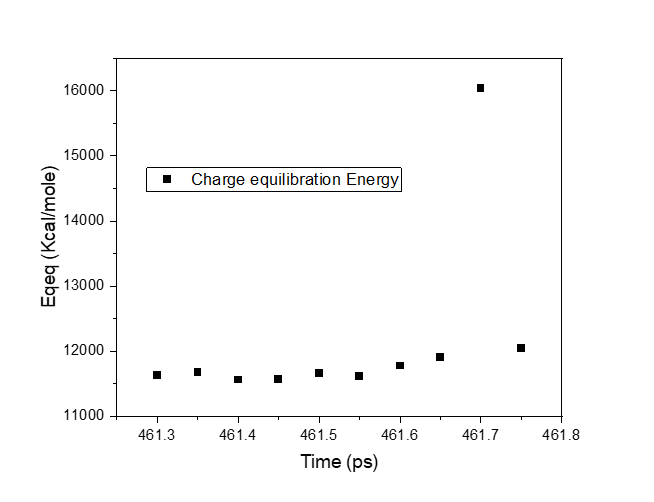
I am thinking therefore, studying why charge equilibration energy showed a surge, might offer an explanation of why the box has exploded.
Do you know what changes to make in charge-related commands such as:
fix 1 all qeq/reaxff 1 0.0 10.0 1.0e-6 reaxff
SO that the box won’t explode?