I am trying to run this simulation:
https://icme.hpc.msstate.edu/mediawiki/index.php/Deformation_of_Amorphous_Polyethylene.html
I receive this error:
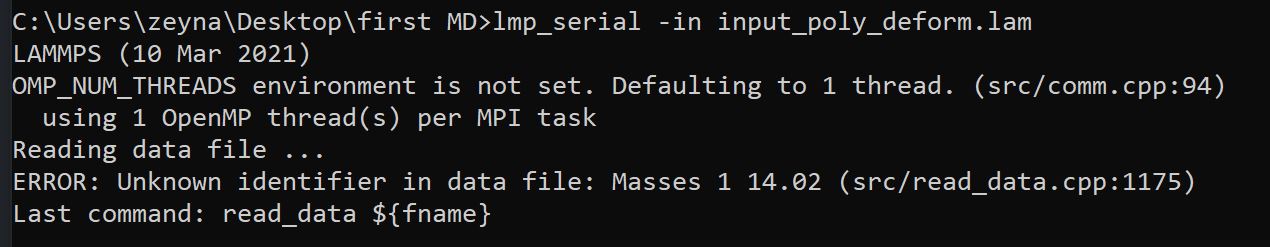
Any help would greatly be appreciated.
I was trying to upload input and data files since I am a new user I could not. the input file is from this page:
https://icme.hpc.msstate.edu/mediawiki/index.php/LAMMPS_Input_Deck_for_Atomistic_Deformation_of_Amorphous_Polyethylene.html
and the data file from here:
https://icme.hpc.msstate.edu/mediawiki/index.php/LAMMPS_Polymer_Datafile.html
Thanks!