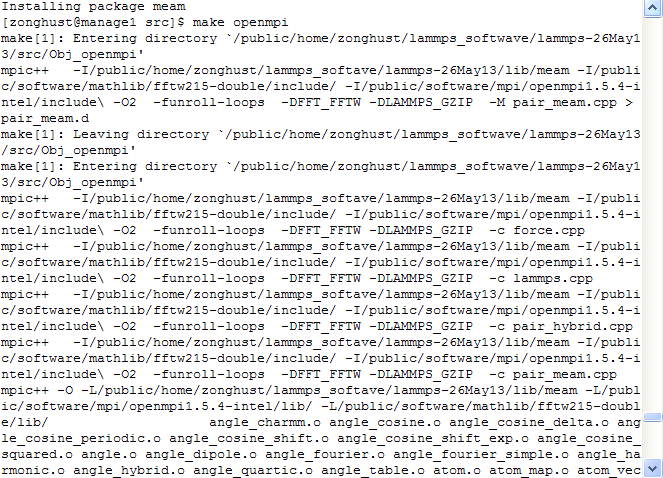
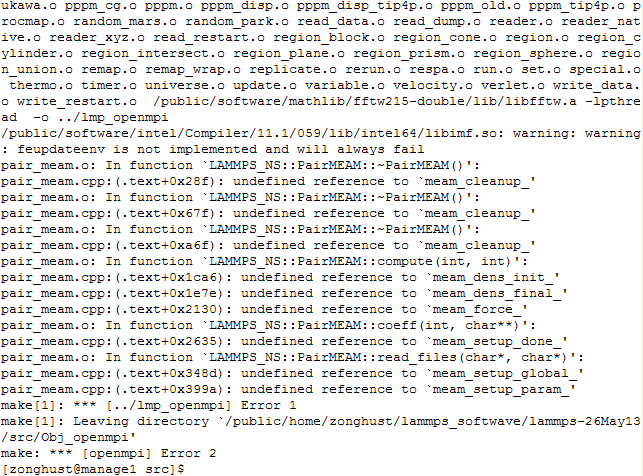
Hi everyone, When i compile meam package in Linux , the same errors always existed. Up to now, i have no idea about the reason and resolutions for those errors. I attached the detials of my operation, hope you can give some advises. (Lammps: 26May13 version), I used fortran and openmpi.
I compile it by following steps: Step: 1) In the directory …/lib/meam— First delete the files of .a, .f, .mod; Then make -f Makefile.gfortran.
get following information:
gfortran O -fpic - fno-second-undersocre c meam_data F。
gfortran O -fpic
fno-second-undersocre cmeam_setup_done F。
gfortran O -fpic
fno-second-undersocre cmeam_setup_global F。
gfortran O -fpic
fno-second-undersocre cmeam_setup_param F。
gfortran O -fpic
fno-second-undersocre cmeam_dens_init F。
gfortran O -fpic
fno-second-undersocre cmeam_dens_final F。
gfortran O
-fpic- fno-second-undersocre cmeam_force F。
gfortran O
-fpic- fno-second-undersocre cmeam_cleanup F。
AR RV libmeam。一meam_data
.Omeam_setup_done.O meam_setup_global.O meam_setup_param.O meam_dens_init.O meam_dens_final.O meam_force.O meam_cleanup.o。
Your errors are occurring at link time, probably b/c you are not including all
the correct libs needed to link a C++ application (LAMMPS) with a Fortran (MEAM)
lib. Which libs are needed is a function of your compilers. Section_start.html
of the manual explains this.
In src/MAKE/Makefile.openmpi you should not need to change anything except
in this section:
compiler/linker settings
specify flags and libraries needed for your compiler
Any Fortran-dependent libs should be added to lib/meam/Makefile.lammps.
Again this is explained in Section_start.html. There are several example
Makefile.lammps.* files i that dir that are useful for different Fortran compilers.
Hi Steve,
Thank you for your replies.
According to your advises, i know some settings should be modified to the makefile.lammps in …/lib/meam file.
The following three settings should be correctly modified.
Followings are my modifications: meam_SYSINC = meam_SYSLIB = -lgfortran meam_SYSPATH = -L/public/software/intel/Compiler/11.1/059/lib/intel64
For meam_SYSPATH , i have no ideas. So, i refers to the following information:
However, there were same errors: eg. undefined reference to " meam_force"
-lgfortran is not an Intel Fortran compiler lib, so those settings
are not consistent. If you compiled with the GNU fortran compiler
than use -lgfortran. If you compiled with the Intel Fortran compiler,
then you need to list Intel Fortran libs for SYSLIB.
See the example Makefile.lammps files in lib/meam. Cases for
both those compilers are there.
Hi Steve,
Following your advice, I modified the compiling of meam package.
I could make it, lmp_openmpi can correctly operate the in.meam file. In the in.meam file, there are 10000 atoms of TiN.
However, i found the calculating speed of linux 48 cores (parallel) is much solwer than that of windows 4 cores (parallel).
eg. for 5000 timesteps, linux needs 2 hrs, while windows only needs less than 1 hrs.
Are this speed dofference related to the settings of compiling files, like makefile.ifort and makefile.openmpi?
Followings are my settings of those two files.
1)make -f Makefile.ifort
2)make yes-meam
3)make openmpi.
If both versions ran, I doubt that the compilation
details are the issue. What do you get
if you run the Linux version on 4 cores, same
as the Windows? Just for a couple 100 timesteps.
Can you post both log files. Are you sure that
both versions are actually running in parallel?
Hi everyone,
There maybe a stupid question to someone. However, it confuses me.
compute 1 precipitate voronoi/atom surface all
compute 2 precipitate reduce sum c_1[3]
Using those two commands, we can know the surface area of the “precipitate” group (the
hull of the precipitate) .
So my confusion is about units of the surface area.
According to the “units” command.
i think the units of the surface area could be like this.
Metal: A2
LJ: sigma2
real: A*2 yes or not?
Hai-Long Yao
State Key Laboratory for Mechanical Behavior of Materials
School of Materials Science and Engineering, Xi’an Jiaotong University
Xianning West Road 28, Xi’an, Shaanxi, 710049 China
Tel: +86 29 82665299
Fax: +86 29 83237910
E-mail:yhl206@…127…
Hi daniel,
There maybe a stupid question to someone. However, it confuses me.
compute 1 precipitate voronoi/atom surface all
compute 2 precipitate reduce sum c_1[3]
Using those two commands, we can know the surface area of the “precipitate” group (the
hull of the precipitate) .
So my confusion is about units of the surface area.
According to the “units” command.
i think the units of the surface area could be like this.
Metal: A2
LJ: sigma2
real: A*2 yes or not?
Another confusion :
If the precipitate group is crushed into several parts, it means the surface area of the precipitate group increases.
In this case, can the “compute voronoi/atom” command correctly calculate the surface area of the precipitate group (including several parts)?
Regardes
Hai-Long Yao
State Key Laboratory for Mechanical Behavior of Materials
School of Materials Science and Engineering, Xi’an Jiaotong University
Xianning West Road 28, Xi’an, Shaanxi, 710049 China
Tel: +86 29 82665299
Fax: +86 29 83237910
E-mail:yhl206@…127…
compute 2 precipitate reduce sum c_1[3]
Using those two commands, we can know the surface area of the “precipitate” group (the
hull of the precipitate) .
No! These commands do not compute the hull! If “surface all” is specified the internal voronoi faces are counted as well. You need to create a “matrix” group, which consists of all-precipitate and specify “surface matrix”.
Dear steve and all,
From most published papers,a modified charge transfer-embedded atom method
potential can be used for simulations of metal-metal oxide systems( eg. Al-O).
However, i cannot find a responding pair-style in lammps. Is this potential not implemeted in lammps?
If yes, i wonder how to make it.
Best regards.
Hai-Long Yao
State Key Laboratory for Mechanical Behavior of Materials
School of Materials Science and Engineering, Xi’an Jiaotong University
Xianning West Road 28, Xi’an, Shaanxi, 710049 China
Tel: +86 29 82665299
Fax: +86 29 83237910
E-mail:yhl206@…127…
Dear Shan,
Thank you for your reply.
I intend to simulate the Al-O system. Althrough “MODIFIED EMBEDDED ATOM METHOD CALCULATIONS OF INTERFACES” gave some
parameters for Al-O interaction in MEAM potential, i think there are not enough. So, i wonder to know and use the Streitz-Mintmire potential.
According to 'MODIFIED EMBEDDED ATOM METHOD CALCULATIONS OF INTERFACES‘,the Al-O.meam file is following:
lattce(1,2)=‘b1’
Ec(1,2) = 4.000
alpha(1,2) = 4.5
re(1,2) = 1.97
re(1,1) = 2.86
re(2,2) = 1.21
rc = 4.0
delr = 0.1
bkgd_dyn=1
Maybe lack of parameters of Cmin(i,j,k) and Cmax(i,j,k) induce the useless of the Al-O.meam file
Could you give some advice about the Cmin(i,j,k) and Cmax(i,j,k)?
Best regards
It’s a pity for such a famous potential. I meam the potential developped by Baskes MI.
Some papers (eg.Attachment ) used the meam potential for Al_Al2O3 interface and the their references are “Baskes MI. Modified embedded atom method calculations of interfaces. Sandia 1996;96:8484.” and "M.I. Baskes, Modified embedded atom method calculations of interfaces, in: S.
Nishijima, H. Onodera (Eds.), Sandia National Laboratories Livermore, USA,1996.".
However, they didn’t show the complete parameters for the Al-O.meam file.
Can anyone give hints excepting follows ?
I don’t understand your comment of “a pity for such a famous potential”…
Anyway, Streitz-Mintmire and MEAM are not related and using one does not necessarily require another.
Your subject title refers to S-M, but you are really asking a question about MEAM. I suggest you open a new thread with a proper title to get maximum attention.