Dear All,
I was trying to calculate a surfactant/water system on a Al2O3 surface.
The simulation box was set up with packmol code, where I placed the Al2O3 at the bottom of a box and then inserted a certain number of surfactant (SDS) and H2O molecules. Since the Al2O3 will be treated rigid, I thought the X and Y lengths of the simulation box are also fixed. In order to make the packing process easier, the Z length was intentionally a larger number (162.5 angstrom instead of the actual 112.5 angstrom, which is estimated from the number of SDS/H2O and their density at such T, P condition).
Once the initial configuration file was prepared, I though the ‘fix npt’ will bring the volume to the correct value. So I am expecting that the X and Y lengths of the box be fixed (due to a rigid Al2O3 surface model) and the Z length will be shrinking from 162.5 angstrom to roughly 112.5 angstrom. Of course the final Z value might be a bit different, because the SDS micellization.
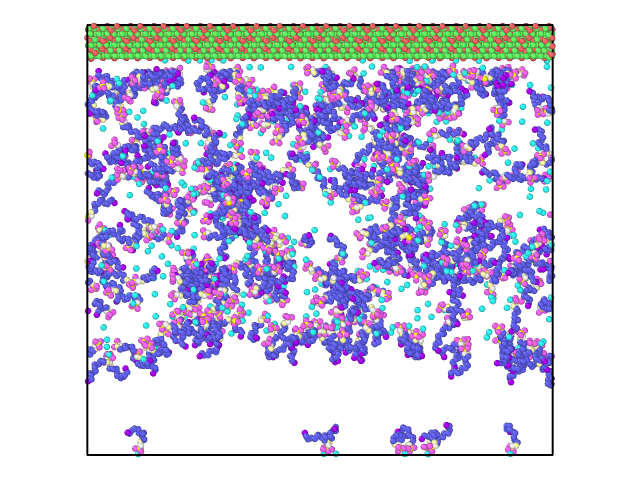
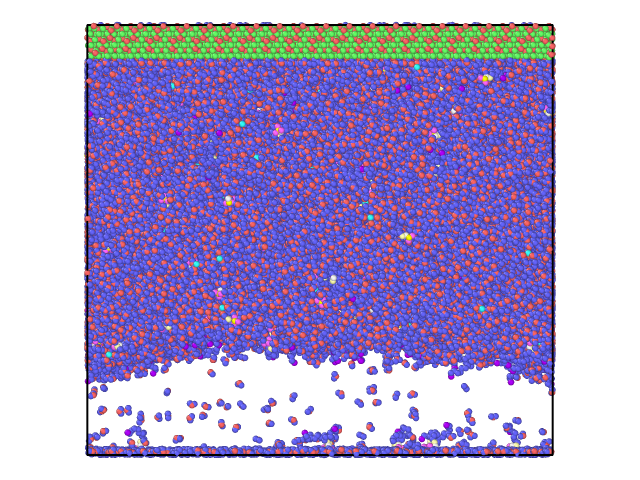
However, this was not I observed. With a few steps, the Z length was expanded to be around 172 angstrom and stayed there for the following 130000 steps (time step 1.0 fs). There is a vacuum like space with few SDS or H2O molecules, see attached to snapshots at step 130000.
I did monitor some other values: the X, Y lengths of the simulation box are not changing during the test run; the center of mass of the Al2O3 is not shifting; the temperature is quite nicely equilibrated around the targeted value; the pressure is fluctuating +/- 200.0 around the setup value 1.0atm, implying the pressure control was not very successful yet.
But what brothers me is, why this was happening? Why the ‘fix npt’ was not able to reduce the volume (the Z length) to a reasonable number? Shall I keep it running for much longer and hope it works? Or I am missing any key control? To me, this looks like both the Al2O3 surface and its mirror images are fixed, preventing the volume from shrinking. Is this what was happening?
The in file is attached. The core part is explained below:
---- core lines from the in file --------
This is to define the groups
group sds type 2 3 4 5 6 7
group na type 8
group al2o3 type 1 9
group water type 10 11
group mobile union sds na water
velocity mobile create 300.0 293288 dist gaussian mom yes rot yes
neighbor 0.3 bin
neigh_modify delay 0 every 1 check yes
neigh_modify exclude molecule al2o3
CFG, Oxygen of H2O is colored as Nitrogen purposely;
dump 3 all cfg 1000 cfg.*.cfg mass type xs ys zs vx vy vz fx fy fz
dump_modify 3 element Al C C O S O C Na O N H
dump_modify 3 sort id
compute tempFree mobile temp
compute pressFree all pressure tempFree
thermo 200
thermo_style custom step vol c_tempFree c_pressFree temp press
thermo_modify line one
fix 1 water shake 0.0001 20 0 b 8 a 9
fix 2 mobile npt temp 300.0 300.0 100.0 iso 1.0 1.0 1000.0
fix_modify 3 temp tempFree press pressFree
run 5000000
----- end of the in file --------
I tried both ‘iso’ and ‘aniso’ in the ‘fix npt’ part, no difference.
Also tried to add this command:
fix 1 al2o3 rigid single force * off off off torque * off off off
However, this brings forth a new problem: the simulation box was expanding nonstop along X, Y and Z directions.
I greatly appreciate any comment and thank you in advance for reading this message!
Best wishes,
Paul
—Paul Huang
Chemical Engineering
University of Oklahoma
lmp3.in (4.6 KB)
