when i use body module,at end I use fix all nev/body ,but the total energy is nonconservation of energy
the in file is
the result is
so is this module not well completed?
when i use body module,at end I use fix all nev/body ,but the total energy is nonconservation of energy
the in file is
the result is
so is this module not well completed?
when i use body module,at end I use fix all nev/body ,but the total energy
there is no good reason to use the body package unless you want to
learn how to use it for programming additional body styles. your
message/question suggests, that this is not your desire. so please
explain why you are trying to use this style and not one of the fix
rigid fixes and regular atom styles? you seem to be making your life
needlessly complicated.
is nonconservation of energy
the in file is
please don't use screen shots, but actual text files: screen shots
waste space and they cannot be commented on. in fact, people that read
e-mail in text mode (yes, those exist; and they are often the more
senior/experienced people) cannot easily see them at all.
the result is
so is this module not well completed?
what do you mean by that?
i didn't see any timestep command in your screen shot. did you check,
that your timestep is adequate? the time integration of the rotational
degrees of freedom for rigid bodies often requires a significantly
shorter timestep than for individual atoms.
axel
thank you very much ,this body module is very important for us ,now we are develop a force feild ,the force feild is using for the rigid body,but it is not
as the rigid body in lammps ,it likes the body module in lammps,we want to use a customized interaction between a pair of bodies,which is not based on sub-particles,for example the water simulation,we take the water as a rigid body ,the integration is based on the center of the mass,the rigid body include translation and rotation,so when i use the body module,i want to test whether the module is useful.and chose the nve. i alse debug the source code ,i am a computer programmer,but the total energy is nonconservation,i debug the source code,the temperture of body is different with temp,
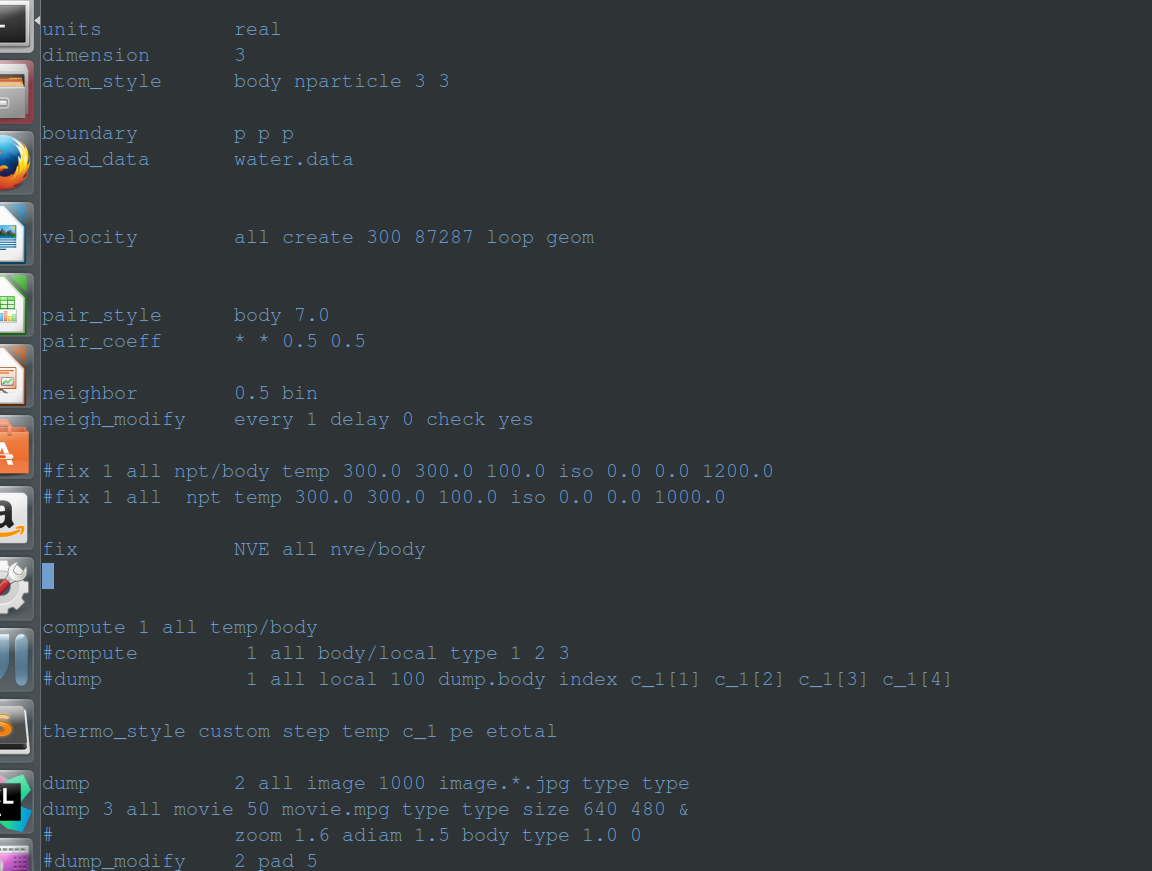
the in file is
units real
dimension 3
atom_style body nparticle 3 3
boundary p p p
read_data water.data
velocity all create 300 87287 loop geom
pair_style body 7.0
pair_coeff * * 0.5 0.5
neighbor 0.5 bin
neigh_modify every 1 delay 0 check yes
#fix 1 all npt/body temp 300.0 300.0 100.0 iso 0.0 0.0 1200.0
#fix 1 all npt temp 300.0 300.0 100.0 iso 0.0 0.0 1000.0
fix NVE all nve/body
#fix 1 all nvt/body temp 1.44 1.44 1.0
#fix 2 all enforce2d
compute 1 all temp/body
#compute 1 all body/local type 1 2 3
#dump 1 all local 100 dump.body index c_1[1] c_1[2] c_1[3] c_1[4]
thermo_style custom step temp c_1 pe etotal
dump 2 all image 1000 image.*.jpg type type
dump 3 all movie 50 movie.mpg type type size 640 480 &
#dump_modify 2 pad 5
timestep 0.001
thermo 100
run 100000
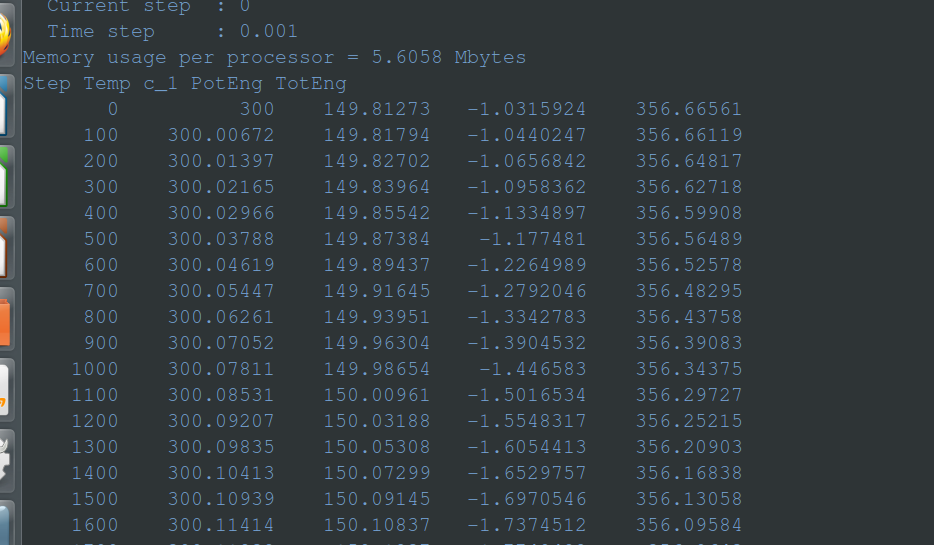
but the result is
Memory usage per processor = 5.6058 Mbytes
Step Temp c_1 PotEng TotEng
0 300 149.81273 -1.0315924 356.66561
100 300.00672 149.81794 -1.0440247 356.66119
200 300.01397 149.82702 -1.0656842 356.64817
300 300.02165 149.83964 -1.0958362 356.62718
400 300.02966 149.85542 -1.1334897 356.59908
500 300.03788 149.87384 -1.177481 356.56489
600 300.04619 149.89437 -1.2264989 356.52578
700 300.05447 149.91645 -1.2792046 356.48295
800 300.06261 149.93951 -1.3342783 356.43758
900 300.07052 149.96304 -1.3904532 356.39083
1000 300.07811 149.98654 -1.446583 356.34375
1100 300.08531 150.00961 -1.5016534 356.29727
1200 300.09207 150.03188 -1.5548317 356.25215
1300 300.09835 150.05308 -1.6054413 356.20903
1400 300.10413 150.07299 -1.6529757 356.16838
1500 300.10939 150.09145 -1.6970546 356.13058
1600 300.11414 150.10837 -1.7374512 356.09584
1700 300.11838 150.1237 -1.7740482 356.0643
1800 300.12213 150.13743 -1.8068319 356.03599
1900 300.12541 150.14959 -1.8358758 356.01086
2000 300.12825 150.16023 -1.8612792 355.98884
2100 300.13066 150.16942 -1.8832199 355.96978
2200 300.13269 150.17724 -1.9018868 355.95353
2300 300.13436 150.18378 -1.9175222 355.93989
2400 300.13571 150.18915 -1.9303397 355.92867
2500 300.13675 150.19344 -1.940575 355.91968
2600 300.13753 150.19675 -1.9484699 355.91271
2700 300.13806 150.19916 -1.9542386 355.90758
2800 300.13837 150.20078 -1.9580966 355.90409
2900 300.13848 150.20168 -1.9602563 355.90207
3000 300.13843 150.20195 -1.9609011 355.90136
3100 300.13822 150.20166 -1.9602051 355.9018
3200 300.13787 150.20087 -1.9583334 355.90326
3300 300.13741 150.19966 -1.9554411 355.9056
3400 300.13684 150.19807 -1.9516445 355.90872
3500 300.13619 150.19616 -1.9470832 355.91251
3600 300.13546 150.19398 -1.9418625 355.91686
3700 300.13466 150.19155 -1.9360755 355.92169
3800 300.13381 150.18894 -1.929822 355.92693
3900 300.13292 150.18615 -1.9231724 355.93251
4000 300.13198 150.18323 -1.9162067 355.93837
4100 300.13102 150.18021 -1.9089789 355.94444
…
i debug the code ,and find that the total energy maybe not the total energy of body. i use 401 body but
the temperture->dof is 1200,and the body dof is 6,should it be 2406?
and c_1 include energy of tranlation and rotation,but the c_1 is half of temp which just include translation
thermo_style does not print out the correct total energy,
and i use 0.5c_12406*boltz+potential energy to compute ,i found the total energy is almost conservation.
maybe the in file i create is wrong ,but now i have a question ,where am i wrong ,
in the future, please have your e-mails to this (english language)
mailing list proofread by somebody that has a better command of
english grammar. i find it difficult to understand the meaning of what
you are trying to say and most likely, i am misunderstanding
something.
thank you very much ,this body module is very important for us ,now we are
develop a force feild ,the force feild is using for the rigid body,but it is
not
as the rigid body in lammps ,it likes the body module in lammps,we want to
use a customized interaction between a pair of bodies,which is not based on
sub-particles,for example the water simulation,we take the water as a rigid
body ,the integration is based on the center of the mass,the rigid body
include translation and rotation,so when i use the body module,i want to
you are not making making sense here. on one hand you talk about
having a model that is different from a rigid body integrator, but
then what you are describing is *exactly* what the rigid body
integrators are doing.
test whether the module is useful.and chose the nve. i alse debug the source
as i mentioned in a previous mail. if you develop a force field, it
should not matter what integrator you are using.
code ,i am a computer programmer,but the total energy is nonconservation,i
debug the source code,the temperture of body is different with temp,
these all sound like random statements, that don't seem to follow any
logic of reasoning.
what has the fact that you are "a computer programmer" to do with
energy conservation.
you twice say "i debug the source code", but what does kind of meaning
does the contribute to your statement?
the in file is
# 2d polygon nparticle bodies
units real
dimension 3
atom_style body nparticle 3 3boundary p p p
read_data water.datavelocity all create 300 87287 loop geom
pair_style body 7.0
pair_coeff * * 0.5 0.5
these are very strange parameters for lennard jones interactions of
atoms in real units. also, typical 3-site water potentials have
charges, which are not supported by the nparticle body style.
neighbor 0.5 bin
neigh_modify every 1 delay 0 check yes#fix 1 all npt/body temp 300.0 300.0 100.0 iso 0.0 0.0 1200.0
#fix 1 all npt temp 300.0 300.0 100.0 iso 0.0 0.0 1000.0
fix NVE all nve/body
#fix 1 all nvt/body temp 1.44 1.44 1.0
#fix 2 all enforce2dcompute 1 all temp/body
#compute 1 all body/local type 1 2 3
#dump 1 all local 100 dump.body index c_1[1] c_1[2] c_1[3] c_1[4]thermo_style custom step temp c_1 pe etotal
dump 2 all image 1000 image.*.jpg type type
dump 3 all movie 50 movie.mpg type type size 640 480 &
# zoom 1.6 adiam 1.5 body type 1.0 0
#dump_modify 2 pad 5
timestep 0.001thermo 100
run 100000but the result is
Memory usage per processor = 5.6058 Mbytes
Step Temp c_1 PotEng TotEng
0 300 149.81273 -1.0315924 356.66561
100 300.00672 149.81794 -1.0440247 356.66119
[...]
i debug the code ,and find that the total energy maybe not the total energy
of body. i use 401 body but
the temperture->dof is 1200,and the body dof is 6,should it be 2406?
which "temperature->dof" in which class instance are you referring to here?
actually, total DOF for the whole system with 401 rigid bodies should be 2403.
1200 would be the number for a standard temperature compute (3*401-3),
which assumes point particles e.g. the one defined by default for
thermo output. this can be changed using thermo_modify.
and c_1 include energy of tranlation and rotation,but the c_1 is half of
temp which just include translation
thermo_style does not print out the correct total energy,
and i use 0.5*c_1*2406*boltz+potential energy to compute ,i found the total
energy is almost conservation.
maybe the in file i create is wrong ,but now i have a question ,where am i
wrong ,
this is another difficult to understand blob of text. the way i see
it, compute c_1 computes temperature correctly (~double the DOFs vs.
compute temp => ~ half the temperature).
my recommendation is that rather than digging around in some
(currently quite experimental) code that you don't seem to understand
while struggling with using LAMMPS overall, you should first build an
input for something simpler and see if you can reproduce published
results.
how about some well known water potentials, e.g. TIP3P. while this is
usually held rigid with SHAKE or RATTLE+SHAKE (for computational
efficiency), you can also use it with fix rigid. making this work (and
comparing thermodynamic data and structural properties with published
results and results from using fix shake / fix rattle) should give you
a better handle at how to adjust LAMMPS input parameters to retain
proper energy conservation.
axel.