Hi, Lammps users,
A weird result: coulombic force stop atom moving in Lammps. I tried several ways to check what’s wrong: atom overlap at periodic boundary; force parameters setup-pair_style; K_space solver, plot of system temp, press,Potential E, Kinetic E. I am confused about the results.
The system are composed of 1000 SPC/E water above a solid wall MgO, T=300k, NVT, water use shake constraint; wall is rigid; dt=3fs; PPPM 0.0001 for k_space. Domain size is Lx=Ly=Lz=100.8 Angstrom. I even tried larged domain size Lx=Ly=Lz=120.8 Angstrom.
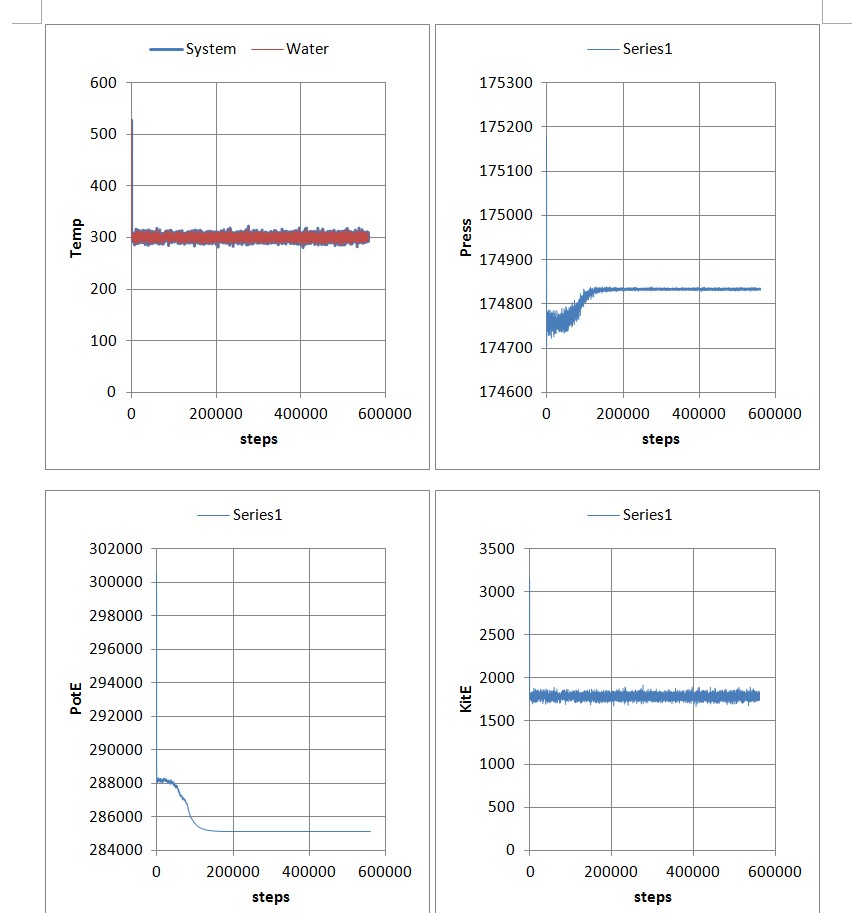
When i charge the wall atoms, the water molecules will become stationary after about 900ps. I put a movie (a period when water molecules obviously going stationary) and plots which u can see. Also the input file and Geometry dat file.
If no charge to the wall atoms, the water molecules move like normal.
My concern is if no atoms move at all: according to equation: sigma_(0.5mivi^2)=1.5NKb*T, Temp should be close to zero. But in plot the water temp is about 300K. But from movie we can see almost not water molecule move at end if wall are charged.
I can not think out some reason for such weird results. Is it possible the Lammps did not install correctly for some reason?
Any comments?
Sincerely,
Yanfei
in.Ruf0Q5Bas48 (3.56 KB)