Dear Axel,
I am writing after a year, having spent long hours working with LAMMPS. I have two points.
- You said in your previous email:
“you are the first person in almost 10 years that i recall being confused in this way by what is in the docs about the DPD pair style.”
Given you are well-known for your passive-aggressive responses, I can only guess how keen people are to write if they are confused about something in LAMMPS. There might be some bias, like there were no complaints about human right abuses in Stalinist Russia.
More importantly, in December 2015 I wrote an email about my confusion with thermostatting DPD, and you responded: “i have to admit that the pair style dpd documentation is lacking a bit in this regard.” I find these two claims inconsistent. Why is it surprising that I am confused, if you yourself admit that the documentation is lacking?
i don’t think that those two statements are in contradiction. lots of people have used LAMMPS DPD (and there are now lots of more elaborate versions of DPD available as well). since nobody there has not been any complaint for such a long time, i believe i can assume, that for all of those people the docs were sufficient.
also, in those two e-mails your are confused about two very different things. and the behavior you are describing in your december 2015 e-mail is not unusual. the questions you ask are those of a person with limited experience in MD fundamentals.
Hence, I suggest adding the attached file dpd.in to the DPD page of the manual as a standard example of how to simulate water, or any single-component liquid. At least the critical lines of it. (The use of SI units was advised to me by Sergey Litvinov, I think LJ units would be as good). I am sure many readers would appreciate that.
- A year ago, I mentioned in this thread that simulating polymers in LAMMPS fais to produce correct results, according to standard literature (Groot, JCP, 1998). Recently, I have spent a lot of time investigating this problem, which is the reason I am writing again. I have thoroughly compared the results from LAMMPS with DL_MESO (denoting DLMS), another package with implemented DPD.
I have done the following runs, with like species interaction parameters “a” and unlike “a+da”, timestep dt=0.05, 10k steps:
- DLMS, binary mixture, a=25, da=10, correct result, phase separation visible in VMD,
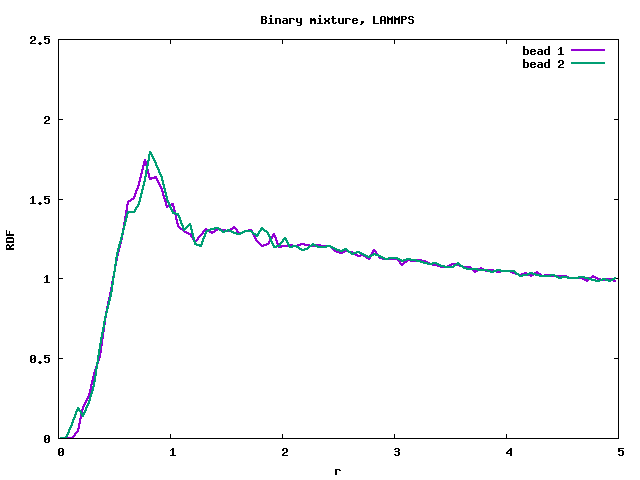
- LMP, binary mixture, a=25, da=10, correct result, phase separation (attached input file binmixt.in),
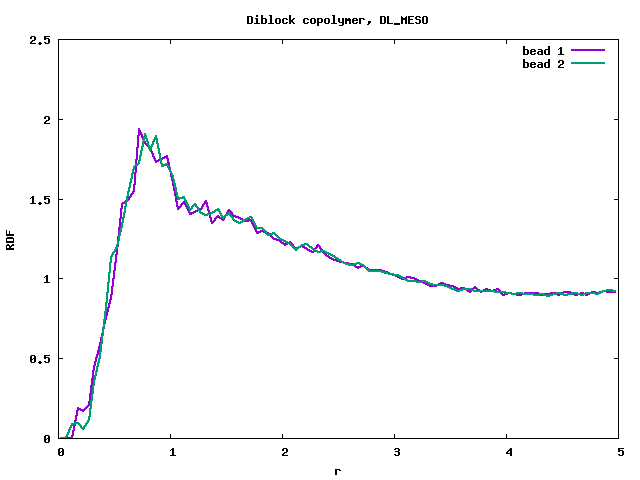
- DLMS, diblock copolymer, a=25, da=10, correct result, lamellar phase established,
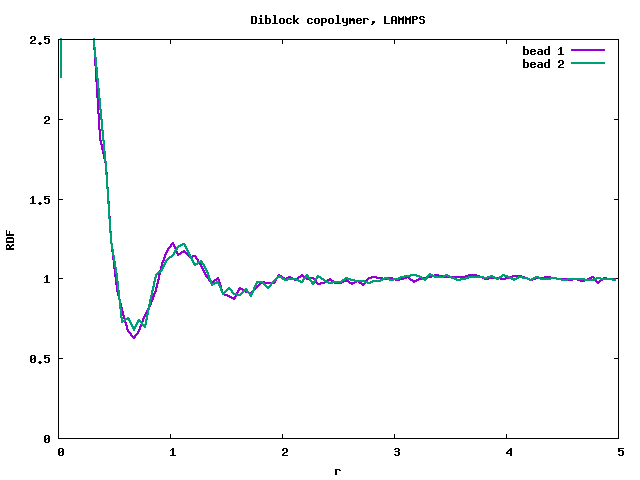
- LMP, diblock copolymer, a=25, da=10, wrong result, no phase separation (attached input file diblock.in).
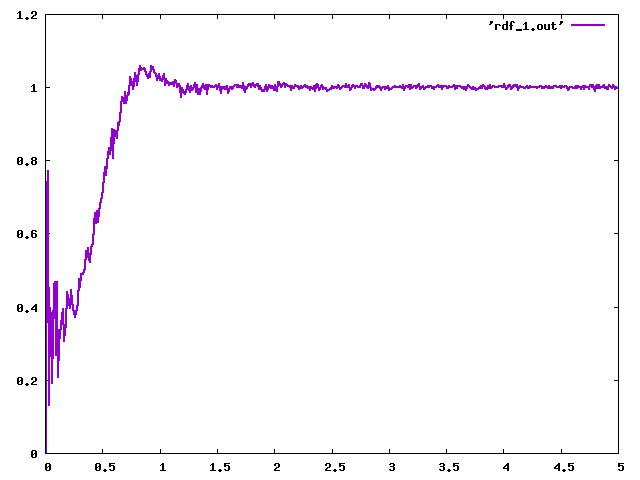
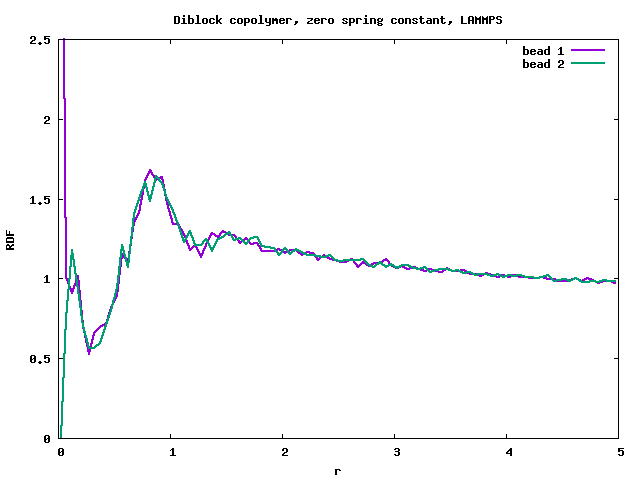
Each time I monitored the radial distribution function (RDF), which can reveal many problems.
It is visible that the RDF from LAMMPS (rdf_lmp_diblock.png) produces a massive peak at r=0, which is a very strange behaviour. Compare with DLMS (rdf_dlms_diblock.png).
I tested a limiting case, setting “bond_style harmonic” to 0 0. The polymer melt with freely moving beads is now essentially a binary mixture (for correct behaviour, see rdf_lmp_binmixt.png). In this case VMD showed a clear phase separation, but the RDF is again wrong, with a peak at r=0 (see rdf_lmp_diblock_zero_spring.png).
At this point, the only difference in the input files between binary mixture (binmixt.in) and diblock copolymer with zero spring constant is “atom_style atomic” vs “atom_style molecular”/“atom_style bond”. Clearly, redefining the atom_style changes physics. Here I am really confused and not sure how I can proceed further to squeeze the correct behaviour from LAMMPS, which is the reason I am writing again.
having bonds with no force constant and having no bonds is resulting in different behavior due to “exclusions”. even if there are no forces from the bonds, the corresponding pairs of atoms are excluded from the neighbor list. you also have to adjust the exclusions settings via the “special_bonds” command as well to make those two settings to represent the exact same physics. again, you are showing lack of experience in MD fundamentals, and thus are not adding much credibility to your findings.
I believe this is strong enough case for you to take notice. I fully realise that LAMMPS is a free open-source code with no warranty and you are in no way obliged to solve my problems. But I think you might be interested.
no, i am not. you have to do a better job to provide convincing evidence. the statistics are stacked against you. very experienced people have used DPD in LAMMPS and while it is not impossible, that there is a bug, it is extremely unlikely, and you are not providing any convincing evidence. if you get different results with a different software, it may just be, that that software has different default settings in parameters, that you don’t set explicitly (e.g. exclusions) and don’t check for.
axel.