Dear LAMMPS users,
I am working on the formation of defects in Fe under external irradiation.
My simulation box is defined a bit larger than the crystal in the z dimension (negative values) so I can define the projectile with its initial position (and velocity) outside the crystal.
boundary p p p
Definition of the simulation box
region simdom block 0. {ncellx} 0. {ncelly} -10. ${ncellz}
Definition of the Fe material
region crystal block 0. {ncellx} 0. {ncelly} 0. ${ncellz}
create_box 2 simdom
create_atoms 1 region crystal
The boundary conditions are periodic in all dimensions.
Since I am interested in the formation of interstitials and vacancies, I am using the Voronoi package.
My problem is the following:
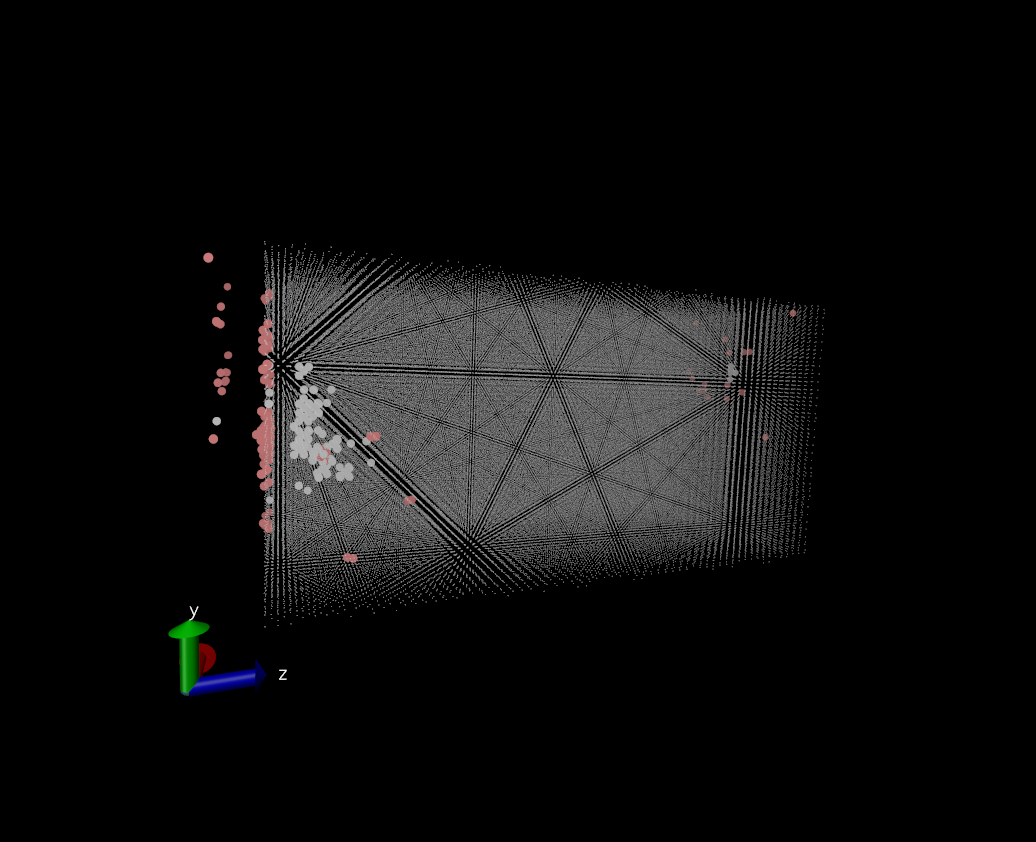
During the impact of the projectile on the surface some atoms of the surface are ejected (towards negative values of z). Since boundary conditions are periodic in z, these atoms cross the boundary and interact with the back side of the crystal. Then, when I calculate interstitials and vacancies that have formed, I can see interstitials and vacancies on the backside, which of course is an artefact (see figure in attachment).
In order to solve that, I tried with ‘s’ boundary conditions in z but I got an error from LAMMPs after some timesteps. Clearly, it is due to the Voronoi package because if I do not use it, the simulation goes smooth.
With periodic BC, there is no problem with Voronoi. But, if I use periodic BC, how could I avoid that atoms that are ejected from the surface cross the boundary and interact with the back side ?
For the moment the idea I have is to compute the z coordinates of each atom and determine which ones have z < 0. But then, once this is done, would it be possible to stop those atoms ?
Many thanks in advance for your help,
Christophe
Dear LAMMPS users,
I am working on the formation of defects in Fe under external irradiation.
My simulation box is defined a bit larger than the crystal in the z
dimension (negative values) so I can define the projectile with its initial
position (and velocity) outside the crystal.
boundary p p p
# Definition of the simulation box
region simdom block 0. \{ncellx\} 0\. {ncelly} -10. ${ncellz}
# Definition of the Fe material
region crystal block 0. \{ncellx\} 0\. {ncelly} 0. ${ncellz}
create_box 2 simdom
create_atoms 1 region crystal
The boundary conditions are periodic in all dimensions.
Since I am interested in the formation of interstitials and vacancies, I am
using the Voronoi package.
My problem is the following:
During the impact of the projectile on the surface some atoms of the surface
are ejected (towards negative values of z). Since boundary conditions are
periodic in z, these atoms cross the boundary and interact with the back
side of the crystal. Then, when I calculate interstitials and vacancies that
have formed, I can see interstitials and vacancies on the backside, which of
course is an artefact (see figure in attachment).
In order to solve that, I tried with 's' boundary conditions in z but I got
an error from LAMMPs after some timesteps. Clearly, it is due to the Voronoi
package because if I do not use it, the simulation goes smooth.
why 's' boundary? why not 'f' boundary and "thermo_modify lost
ignore". that would be the most common approach to avoid artifacts
from those knocked out atoms. they just vanish.
what is the voronoi problem? are you using an up-to-date LAMMPS
binary? there have been multiple updates and bugfixes for the voronoi
compute this year alone.
please have a look at http://lammps.sandia.gov/guidelines.html and
consider the suggestions there when posting to lammps-users.
thanks in advance,
axel.
Dear LAMMPS users,
why ‘s’ boundary? why not ‘f’ boundary and “thermo_modify lost
ignore”. that would be the most common approach to avoid artifacts
from those knocked out atoms. they just vanish.
If I use ‘f’ boundary and “thermo_modify lost ignore flush yes” I still get an error related to lost atoms:
Step Time Atoms Temp kinecpro peproj
1500 13.237689 131073 594.09015 4999.5106 0
1600 13.245298 131073 595.54452 4999.5613 -0.034667001
1700 13.253338 131073 577.83886 201.80982 -0.72538681
1800 13.262942 131073 557.61818 104.08271 2.5752722
1900 13.274639 131073 545.79989 32.185489 1.1045357
2000 13.292804 131073 515.48803 2.3026844 -2.8400814
2100 13.332281 131073 474.54717 0.36001004 -2.9327816
2200 13.397611 131073 423.82665 0.21992154 -3.1066298
ERROR: Lost atoms: original 131073 current 131072 (…/thermo.cpp:398)
I did not use Voronoi for this test. So it might be an issue with LAMMPS and not with Voronoi.
what is the voronoi problem? are you using an up-to-date LAMMPS
binary? there have been multiple updates and bugfixes for the voronoi
compute this year alone.
I am using version 14May2016.
please have a look at http://lammps.sandia.gov/guidelines.html and
consider the suggestions there when posting to lammps-users.
In general I search through the mailing list first. If I do not find the solution to my problem there, I send an email.
Christophe
> Dear LAMMPS users,
>
why 's' boundary? why not 'f' boundary and "thermo_modify lost
ignore". that would be the most common approach to avoid artifacts
from those knocked out atoms. they just vanish.
If I use 'f' boundary and "thermo_modify lost ignore flush yes" I still get
an error related to lost atoms:
Step Time Atoms Temp kinecpro peproj
1500 13.237689 131073 594.09015 4999.5106 0
1600 13.245298 131073 595.54452 4999.5613 -0.034667001
1700 13.253338 131073 577.83886 201.80982 -0.72538681
1800 13.262942 131073 557.61818 104.08271 2.5752722
1900 13.274639 131073 545.79989 32.185489 1.1045357
2000 13.292804 131073 515.48803 2.3026844 -2.8400814
2100 13.332281 131073 474.54717 0.36001004 -2.9327816
2200 13.397611 131073 423.82665 0.21992154 -3.1066298
ERROR: Lost atoms: original 131073 current 131072 (../thermo.cpp:398)
I did not use Voronoi for this test. So it might be an issue with LAMMPS and
not with Voronoi.
that would only happen when you reset your thermo settings *after*
doing thermo_modify, which will invalidate previous settings. please
check your log file carefully. you should see a warning about this.
what is the voronoi problem? are you using an up-to-date LAMMPS
binary? there have been multiple updates and bugfixes for the voronoi
compute this year alone.
I am using version 14May2016.
there are newer versions. you still haven't explained what your
voronoi problem is.
please have a look at http://lammps.sandia.gov/guidelines.html and
consider the suggestions there when posting to lammps-users.
In general I search through the mailing list first. If I do not find the
solution to my problem there, I send an email.
that is only one of the suggestions given there. it is quite tiresome
to have to ask people for the same specific details over and over
again, that should be obvious to anybody following all the points
mentioned in these "10 commandments".
in general, the more specific, concise and complete information you
provide, the easier and less hassle it is to help people. it is always
better to be redundant and more detailed and specific than to be vague
and assuming the other person remembers every detail of previous
discussions? do you have an idea, how many e-mails people like me are
writing day in, day out?
axel.
I did not use Voronoi for this test. So it might be an issue with LAMMPS and
not with Voronoi.
that would only happen when you reset your thermo settings after
doing thermo_modify, which will invalidate previous settings. please
check your log file carefully. you should see a warning about this.
You are right, there was a thermo_style command after thermo_modify. I did not know it could invalidate previous settings. When written after it works properly.
what is the voronoi problem? are you using an up-to-date LAMMPS
binary? there have been multiple updates and bugfixes for the voronoi
compute this year alone.
I am using version 14May2016.
there are newer versions.
Ok, I will install version 30Jul2016, last stable one if I’m not wrong.
you still haven’t explained what your
voronoi problem is.
Well, so far I thought that the problem of lost atoms was generated by Voronoi package. Now it seems it is not the case.
However, I am a bit concerned by the fact that the number of atoms calculated during the first invocation of Voronoi, will not be the same as the one at the end of the simulation since some atoms will be lost.
So far, I was working on cases where the number of atoms is conserved. Voronoi is invoked once at the beginning and then, once at the end of the run. I wrote a script in Python that uses both files and by comparison is able to determine what is an interstitial and what is a vacancy.
Now, the number of atoms changes during the run and I am wondering if this approach is still valid…
please have a look at http://lammps.sandia.gov/guidelines.html and
consider the suggestions there when posting to lammps-users.
In general I search through the mailing list first. If I do not find the
solution to my problem there, I send an email.
that is only one of the suggestions given there. it is quite tiresome
to have to ask people for the same specific details over and over
again, that should be obvious to anybody following all the points
mentioned in these “10 commandments”.
in general, the more specific, concise and complete information you
provide, the easier and less hassle it is to help people. it is always
better to be redundant and more detailed and specific than to be vague
and assuming the other person remembers every detail of previous
discussions? do you have an idea, how many e-mails people like me are
writing day in, day out?
I understand and I apologise. I can imagine how many emails you receive every day…and therefore, I try not to give too many details not to bother you. Thinking I was doing good I did bad. From now on, I will try to be much more specific.
Thanks you again for the help you bring to all of us.
Christophe
[...]
Ok, I will install version 30Jul2016, last stable one if I'm not wrong.
you still haven't explained what your
voronoi problem is.
Well, so far I thought that the problem of lost atoms was generated by
Voronoi package. Now it seems it is not the case.
However, I am a bit concerned by the fact that the number of atoms
calculated during the first invocation of Voronoi, will not be the same as
the one at the end of the simulation since some atoms will be lost.
So far, I was working on cases where the number of atoms is conserved.
Voronoi is invoked once at the beginning and then, once at the end of the
run. I wrote a script in Python that uses both files and by comparison is
able to determine what is an interstitial and what is a vacancy.
Now, the number of atoms changes during the run and I am wondering if this
approach is still valid...
have you looked at the "occupation" feature of compute voronoi/atom?
that sounds like it could be helpful in your case and might simplify
what you want to do.
this is one feature, where i recall LAMMPS developers having corrected
problems due to varying numbers of atoms.
axel.
[…]
Ok, I will install version 30Jul2016, last stable one if I’m not wrong.
you still haven’t explained what your
voronoi problem is.
Well, so far I thought that the problem of lost atoms was generated by
Voronoi package. Now it seems it is not the case.
However, I am a bit concerned by the fact that the number of atoms
calculated during the first invocation of Voronoi, will not be the same as
the one at the end of the simulation since some atoms will be lost.
So far, I was working on cases where the number of atoms is conserved.
Voronoi is invoked once at the beginning and then, once at the end of the
run. I wrote a script in Python that uses both files and by comparison is
able to determine what is an interstitial and what is a vacancy.
Now, the number of atoms changes during the run and I am wondering if this
approach is still valid…
have you looked at the “occupation” feature of compute voronoi/atom?
that sounds like it could be helpful in your case and might simplify
what you want to do.
That is what I use:
INITIAL VORONOI TESSELATION
compute v1 all voronoi/atom occupation
compute r0 all reduce sum c_v1[1]
compute r1 all reduce sum c_v1[2]
thermo_style custom c_r0 c_r1
run 0
Initial coordinates of atoms and tesselation
write_dump all custom coord_atoms_init.xyz id x y z
…
…
run 5000
Final coordinates of atoms and result of occupation from Voronoi
write_dump all custom coord_atoms_final.xyz id x y z c_v1[1] c_v1[2]
Then, by post-processing the data from both files I can identify interstitials and vacancies. I think there is no other way to do that (as far as I know). I think LAMMPS cannot give that directly, right ?
this is one feature, where i recall LAMMPS developers having corrected
problems due to varying numbers of atoms.
I have to modify my Python script and do some tests.
Christophe