Hello everyone! Sorry if I’m making some noise here, but I’ve been
reading the documentation and some things are slightly unclear to
me yet, specifically regarding hybrid potential interactions.
I’m trying to model a set of particles with two different potentials,
but one of them is the sum of two yukawa interactions. Can I do
this with hybrid potentials or should I need to add a specific source
file for it and recompile LAMMPS?
I want atoms to interact with each other with two yukawas, depending
on the species, so the interaction would be:
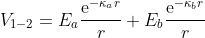
V_{1-2}=E_{a}\frac{\text{e}^{-\kappa_a r}}{r}+E_{b}\frac{\text{e}^{-\kappa_b r}}{r}

V_{1-1}=E_{c}\frac{\text{e}^{-\kappa_c r}}{r}+E_{d}\frac{\text{e}^{-\kappa_d r}}{r}
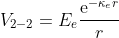
V_{2-2}=E_{e}\frac{\text{e}^{-\kappa_e r}}{r}
but I’m having a hard time realizing how can I accomplish this in the input file.
Has anyone come across this issue before?
Pablo
PS: Sorry if this is already over there in the documentation or the mail-list, but
I couldn’t find it yet.
Yukawa (screened coulomb) is a simple pair potential that can be represented by pair style table for example (with a cut-off). No hybrid potential necessary, just tabulate all the species interactions separately.
Daniel, thanks very much for the quick response. I’ve tried to tabulate
the yukawa potential, but now i’m having some quite serious energy
conservation issues, causing atom losses after every few steps, which
I believe must come from the inner cutoff for the table. Has any of you
ever run into something like this? I’ve tried different ways of making the
table, yet none of them seems to be suitable and I cannot avoid this
atom-loss issue
Pablo
Daniel, thanks very much for the quick response. I've tried to tabulate
the yukawa potential, but now i'm having some quite serious energy
conservation issues, causing atom losses after every few steps, which
I believe must come from the inner cutoff for the table. Has any of you
ever run into something like this? I've tried different ways of making the
it means that atom are getting too close. that may be due to errors in the
tabulation (e.g. wrong sign? wrong units), problems with the initial
structure (did you run a minimization?) or generally bad parameters (time
step, neighbor list settings etc).
table, yet none of them seems to be suitable and I cannot avoid this
atom-loss issue
first you have to find out what is causing the problem. as an alternative
(and check) you can try out using hybrid/overlay, which - as of recent -
allows to add the same potential twice with different parameters on a given
pair of atom types.
axel.
Indeed, that was happening, I created the table with the wrong sign for the force! Sorry for the noise, I’m really ashamed I didn’t realize it.
One last question: suppose I want to know the energy due to each of these terms separately (say, due to the first and the second yukawa in the 1-2 interaction). Could I create each interaction in a different table and overlay them?
Pablo
Indeed, that was happening, I created the table with the wrong sign for
the force! Sorry for the noise, I'm really ashamed I didn't realize it.
don't worry too much about it. you are not the first and won't be the last
to make a mistake like that. the important step is to learn from it.
One last question: suppose I want to know the energy due to each of these
terms separately (say, due to the first and the second yukawa in the 1-2
interaction). Could I create each interaction in a different table and
overlay them?
nope. not directly. this kind of information is not collected separately.
the only way to compute subsets is to first do the simulation and then use
the rerun command on the resulting trajectory, while defining only the
subset of interactions that are of interest.
axel.
Pablo