Dear Users,
I have a system with propane molecules(opls-aa force filed) and a surface. In my system
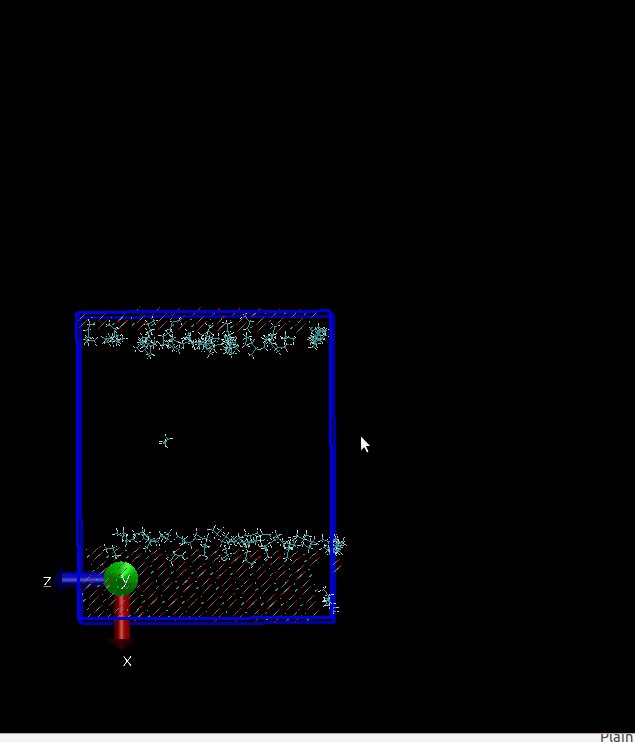
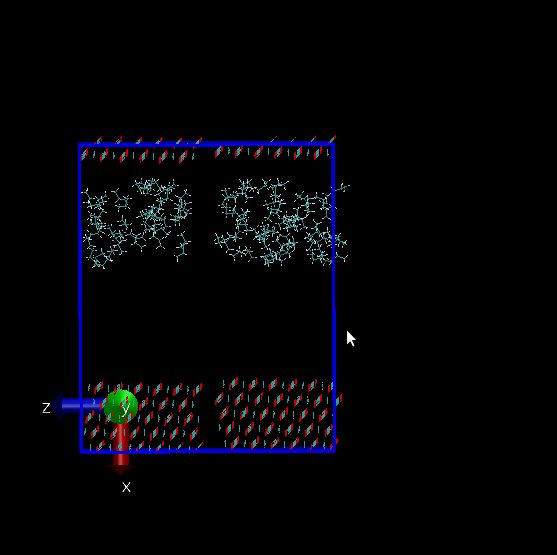
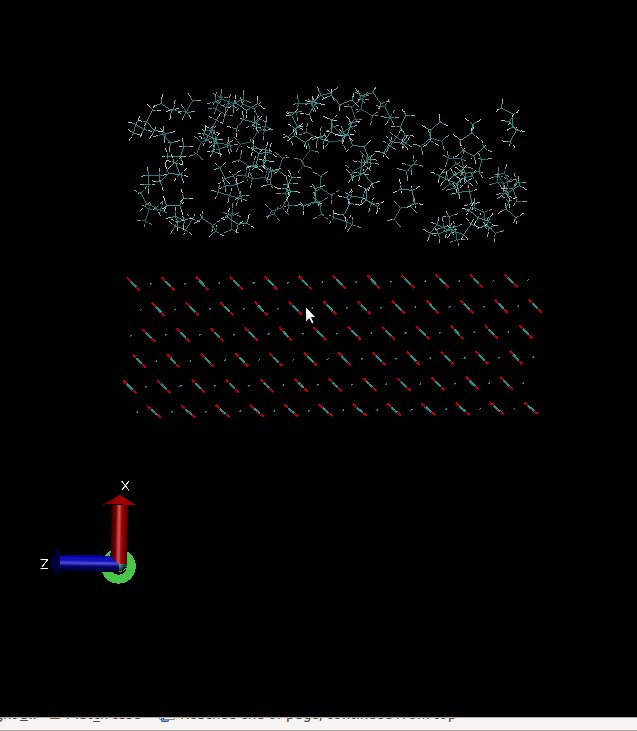
and in x direction at first I have a surface and then above this surface I have propane
molecules. I have a problem, my molecules can sense below my surface(I’ve attach two my
snapshots of initial configuration and final configuration that include gap below my surface but I can not this gap in vmd. I used these two lines for visualizing in vmd :
pbc wrap -compound res -all
pbc box
)
while it should not occur besides I should use pbc in all directions. All of my molecules should
have interaction with above of my surface.
According to my prior email and reply I add extra space by increasing
the length of my box in x direction for having a vacuum slab below my surface. But After running
I got the same results which I have pbc in all directions and without gap below my surface.
Does LAMMPS ignore this vacant room below my surface?
My prior post :
I want to add a gap between my surface and molecular environment. Are ther any options in vmd
for performing this? Or Can I add a gap wherever in my simulation box? I want to omit the
interactions between them, my surface(crystal) and molecules.
In my view one probable approach can be this but I am not sure about it,
My system should be pbc in all directions. I want to add this gap below the end of my
structure therefore when I write the system.data after that I will increase the box size in direction
x while there are not any atoms from the bottom of my system to my structure.
Is it possible? or Will Lammps perform it what I want to reach?
![]()