Hello all,
When I relax my system using NVT time integration, the energy converges but increases. Can I call this a relaxed system? This seems a bit unphysical to me. The code and the plot is attached. Does it have something to do with the damping constant? Some help would be very appreciated.
Saransh
units metal
boundary s s p
atom_style atomic
pair_style eam/alloy
read_data data.au_8_2
pair_coeff * * Au-Grochola-JCP05.eam.alloy Au Au Au Au Au
reset_timestep 0
timestep 0.005
velocity all create 300 134569 mom yes rot no
fix 1 all nvt temp 300.0 300.0 1
variable p11 equal “step”
variable p21 equal “ke”
variable p31 equal “pe”
variable p41 equal “(ke+pe)”
variable p51 equal “temp”
variable p61 equal “press”
fix parameter all print 500 “{p11} {p21} {p31} {p41} {p51} {p61}” file reax_parameter.txt screen no
Set thermo output
thermo 1000
thermo_style custom step lx ly lz press pxx pyy pzz pe temp
dump 1 all atom 2000 dump.*.relax
Run for at least 100 picosecond (assuming 5 fs timestep)
run 20000
unfix 1
unfix parameter
undump 1
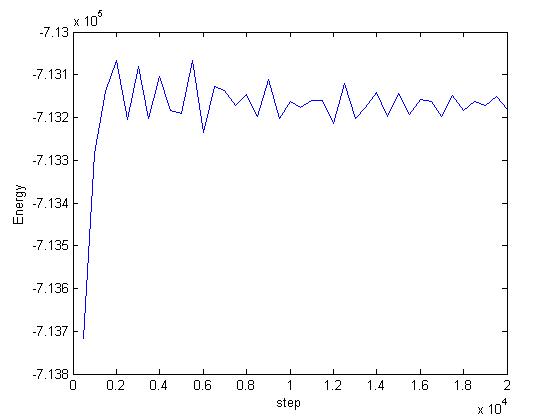
This is absolutely physical. Since the system is thermalized at 300K,
it should have a higher energy.
Ray
When I relax my system using NVT time integration, the energy converges but
increases. Can I call this a relaxed system? This seems a bit unphysical to
me.
If the initial geometry of your system is favorable, then there is no
reason to assume that the energy at t=0 is higher than it would be
when equilibrated at 300K.
Does it have something to do with the damping constant?
fix 1 all nvt temp 300.0 300.0 1
I don't know, but your Tdamp parameter, 1, is smaller than the typical
value. In the online documentation, they recommend using 100.
(http://lammps.sandia.gov/doc/fix_nh.html)
If your goal is to minimize the system, why did you run it at 300K?
try
fix 1 all nvt temp 300.0 0.0 100
(or if that fails, try fix 1 all nvt temp 300.0 0.01 100)
This will cool the system from 300K to zero.
Alternately you can replace the "run" command with the "minimize" command
http://lammps.sandia.gov/doc/minimize.html
Cheers
Andrew
I am trying to get the most stable configuration at 300 K before I start my simulation. A similar system (though larger in size) with exactly the same set of commands yields a different relaxation behavior. I am just trying to understand what might be the reason for this. The plot is attached. Any suggestions or comments are welcome.
Saransh
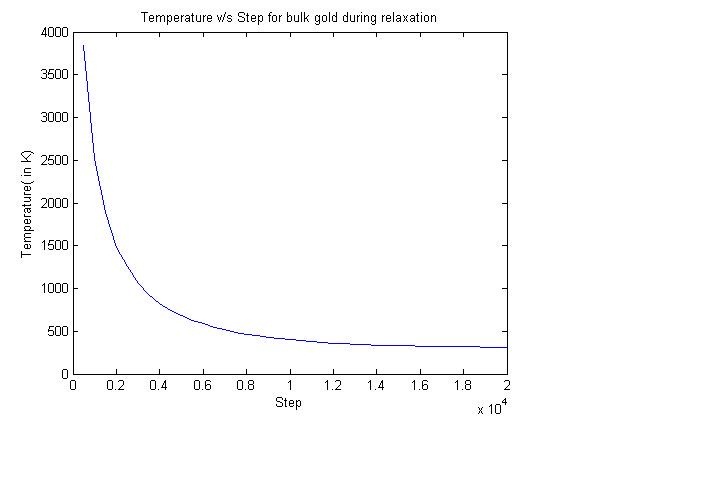
Slight correction. The energy is actually in ev. Not in joules as labeled in the graph
Slight correction. The energy is actually in ev. Not in joules as labeled in
the graph
Frist plot has no label, second plot is labeled with Temperature. I
am not sure what you are referring to.
I am trying to get the most stable configuration at 300 K before I start
my simulation. A similar system (though larger in size) with exactly the
same set of commands yields a different relaxation behavior. I am just
trying to understand what might be the reason for this. The plot is
attached. Any suggestions or comments are welcome.
It maybe because the small system is too small to obtain meaningful statistics.
Ray
Andrew,
The manual states a tdamp of 100 timesteps is appropriate, so that
tdamp should be 100*0.005=0.5.
1 as it was is fine, but 100 is too big.
Ray
oops! My bad. Attached the wrong graph. The smaller system has more that 50,000 atoms. The larger one has about 500,000.
And the previous graph was also energy v/s step.
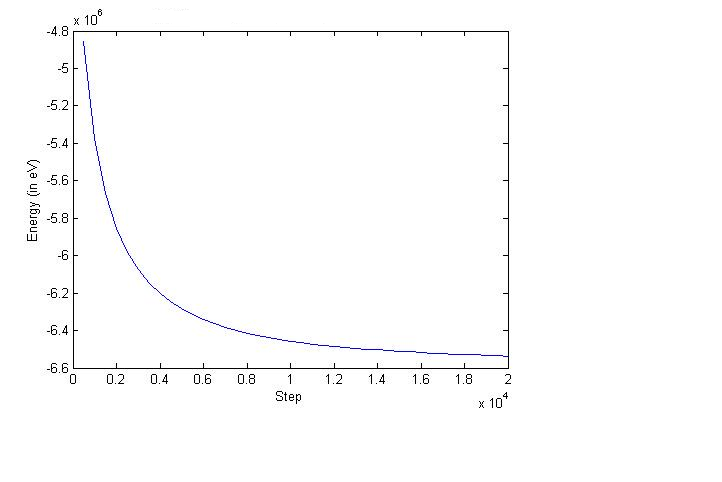
Try compare per-atom energies of the two systems. Also you don't have
periodic boundary conditions so that size effect is more obvious.
Your script looks fine, plots look fine, too. Difference may result
from structures.
Ray
Andrew,
The manual states a tdamp of 100 timesteps is appropriate, so that
tdamp should be 100*0.005=0.5.
1 as it was is fine, but 100 is too big.
I'm quite sorry, Ray is right. (I'm used to using a different unit
system and I was not careful when I posted.) Using a Tdamp of 100
would be bad. I'm glad Ray caught this.
Your script looks fine, plots look fine, too. Difference may
result from structures.
That's what I think too.
Saransh, in your original plot, the energy of the system does not
increase very much per atom (only 0.0012 ev/atom, assuming you have
50000 atoms, which is pretty small compared to kb*T). So it seems
like the magnitude of the energy increase is in the range you could
expect due to thermal equilibration. It just seems that you started
from a favorable initial conformation.
I suspect if you cool the system using:
fix 1 all nvt temp 300.0 0 1
the energy will go down, not up, but try it and let us know.
Andrew