Hi Users,
Could you please provide me help with the following question. I’ve been struggling with it for 3 days now.
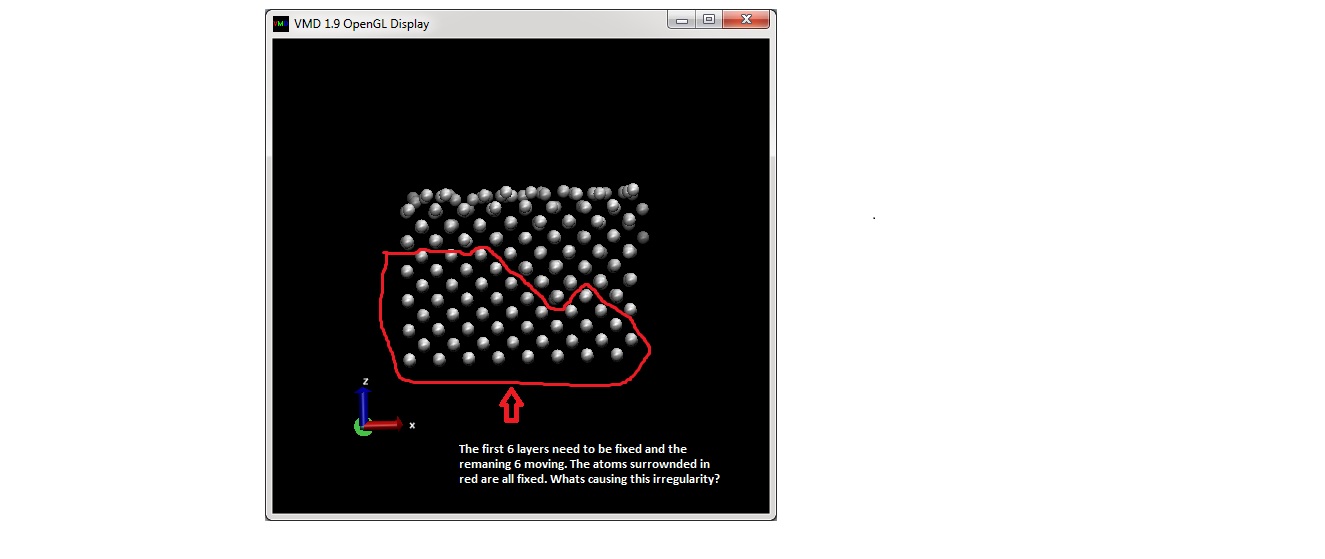
Attached, please find the picture of a VMD run and the input script corresponding to that run. I have 385 atoms and the 385th one is an extra atom which has already been generated. I read an external config file in this case which too is attached. Acc to the input script, I have 192 fixed and 193 moving atoms. Now, when I run the dump, in the Z-direction, 6 layers need to be fixed and 6 moving. The picture attached shows the irregularity in alignment. Whats causing this?
I’d really appreciate the input. Please be as elaborate as possible.
Regards,
Saketh.
Starting.in (1.73 KB)
StartingSi1.xyz (11.1 KB)
Hi Users,
Could you please provide me help with the following question. I’ve been
struggling with it for 3 days now.
Attached, please find the picture of a VMD run and the input script
corresponding to that run. I have 385 atoms and the 385th one is an extra
atom which has already been generated. I read an external config file in
this case which too is attached. Acc to the input script, I have 192 fixed
and 193 moving atoms. Now, when I run the dump, in the Z-direction, 6 layers
need to be fixed and 6 moving. The picture attached shows the irregularity
in alignment. Whats causing this?
what irregularity?
VMD is working fine.
your input is asking for atoms 1 to 192 to be fixed and the rest to be moving.
i have loaded your data file into VMD and marked the two groups in different
colors. they match with what you are showing in your image.
VMD cannot help if your input is not what you think it is.
sorry, but PEBCAC. 
cheers,
axel.
