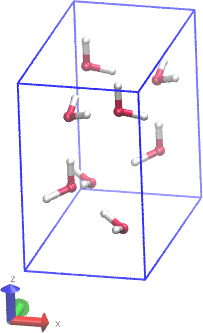
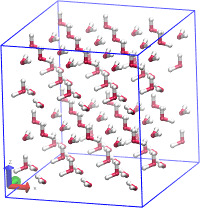
Hi,
I am trying to run a simulation to equilibrate H2O at 1 K in a box that is larger than the periodic boundaries of the system via the “change_box” command so that the box will have enough space for additional atoms. When I extend the box dimensions the system is equilibrating properly with energy but when uploading the coordinates into a visual tool (OVITO) the system is seen to melt. Why is this happening? My input file is below…My ultimate goal is to equilibrate H2O with SiC to calculate the equilibrium properties of the interface between the two structures.
INPUT FILE:
units real
dimension 3
boundary p p p
atom_style full
read_data data.H20.dat
#read_data H2O_648N_Coord.txt #648
#variables-------------------------------------------------------------------------
variable Text equal 1.0
variable Pext equal 0.0
variable Nrun equal 1000000
variable ts equal 0.0016
variable Tdamp equal ${ts}*100
variable Pdamp equal ${ts}*1000
variable Nf equal ${Nrun}/100
variable Ne equal 10
variable Nr equal {Nf}/{Ne}
variable Ndump equal ${Nrun}/2
variable Nr_rdf equal 0.5*{Nrun}/{Ne}
variable watMoleMass equal 18.0153 # /(g/mol)
variable nAvog equal 6.0221415e23 # Avogadro’s number
variable watMoleculeMass equal ({watMoleMass}/{nAvog}) # /(g/molecule)
variable A3_in_cm3 equal 1e-24 # Angstrom^3 in cm^3
variable nAtoms equal atoms
variable nMolecules equal v_nAtoms/3
#create groups ###---------------------------------------------------------
mass 1 1.00794 # H
mass 2 15.9994 # O
#include forcefield.TIP4P-2005.txt
TIP4P/2005 potential parameters-------------------------
group O type 2
group H type 1
#group H2O type 1 2
TIP4P/2005 flexible potenrial parameters---------------------------------
pair_style lj/cut/tip4p/long 2 1 1 1 0.1546 12.0
kspace_style pppm/tip4p 1e-5
pair_coeff 1 1 0.0 0.0
pair_coeff 1 2 1 0.0 1.5795
pair_coeff 2 2 0.1852573718 3.1644
bond_style morse
bond_coeff 1 103.38934 2.287 0.9419
angle_style harmonic
angle_coeff 1 43.95435 107.4
replicate 6 6 6
change_box all z delta -10.0 10.0 x delta -10.0 10.0 y delta -10.0 10.0 units box
neighbor-----------------------------------------------------------------
neighbor 2.0 bin
neigh_modify delay 0 every 1 check yes
#Run one minimization to settle the lattice
fix 1 all nve
fix 2 all box/relax iso 0.0 vmax 0.001
min_style cg
minimize 1.0e-12 1.0e-12 100000 1000000
#print the entire lattice (used for SIA and Vac search) -#but we only wanted printed out now
run 1
#undump startdump
unfix 1
#dumps and restart-----------------------------------------------------------
initial condition------------------------------------------------------
velocity all create ${Text} 1234546 dist gaussian mom yes rot yes
#dump waterdump all custom 1000 lammps.coord_vel id type x y z vx vy vz
dump trj all atom ${Nf} WaterTrj.data
dump_modify trj scale no sort id
dump deqlb all atom ${Nrun} FinalCoord.data
dump_modify deqlb scale no sort id
dump coord all atom ${Ne} DumpCoord.txt
dump_modify coord scale no sort id
timestep ${ts}
fix 4 all npt iso {Pext} {Pext} {Pdamp} temp {Text} {Text} {Tdamp}
variable Dens equal v_nMolecules*{watMoleculeMass}/(vol*{A3_in_cm3})
fix DensAve all ave/time {Ne} {Nr} ${Nf} v_Dens file wat.dens.data
output-------------------------------------------------------------------
thermo_style custom step temp pe ke etotal press vol
thermo_modify flush yes norm yes
thermo ${Nf}
run ${Nrun}
write_restart restart.H2O_16p
Thanks
David Blanton