Dear all,
I using lammps to simulate polymer melts and solutions based on sdpd particles joined by fene springs. I subject these systems to Reverse Poiseuille Flow using
Apply forcing
variable bodyfx atom mass*{gx}*((y>{Ly}/2.0)-(y<${Ly}/2.0))
fix reverse_periodic all addforce v_bodyfx 0.0 0.0
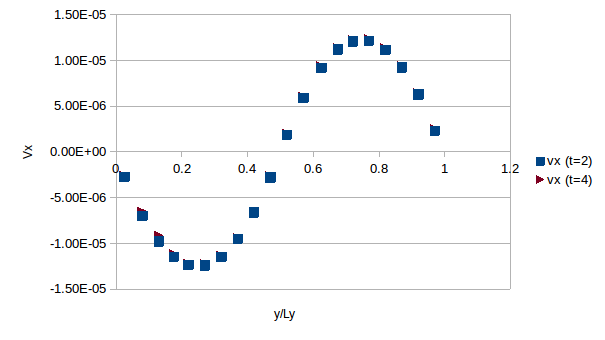
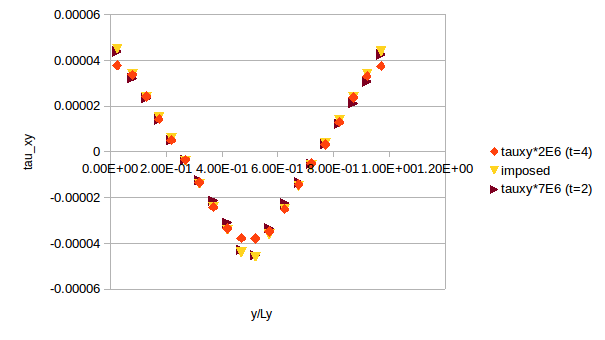
My simulations seem to be running ok since the velocity profile that I obtain for the solvent particles agree with V_y = rhogx/(2eta)(yLy-y^2). For the melts and solutions I obtain reasonable results (i.e., increased viscosity lowers vmax and a bit of flattening of the profiles due to power law-like thinning). Nevertheless when I am computing the stress of the system I find some strange results. For the solvent I find linear profiles that are proportional to tau_xy= rhogxLy/4*(1-4*y/Ly) but not quite correct. The scaling factor seem to depend on the system size.
I calculate the stress by chunking the domain:
compute chunk_vi all chunk/atom bin/1d y lower 0.01 units reduced
and the stress with:
compute stress_v all stress/atom NULL virial
variable sv_xx atom c_stress_v[1]
variable sv_yy atom c_stress_v[2]
variable sv_zz atom c_stress_v[3]
variable sv_xy atom c_stress_v[4]
fix av_stress_v all ave/chunk 1 {Nfreq} {Nfreq} chunk_vi v_sv_xx v_sv_yy v_sv_zz v_sv_xy file sv.av ave window 5
I divide the output stress by the per-atom-volume = vol/count(all).
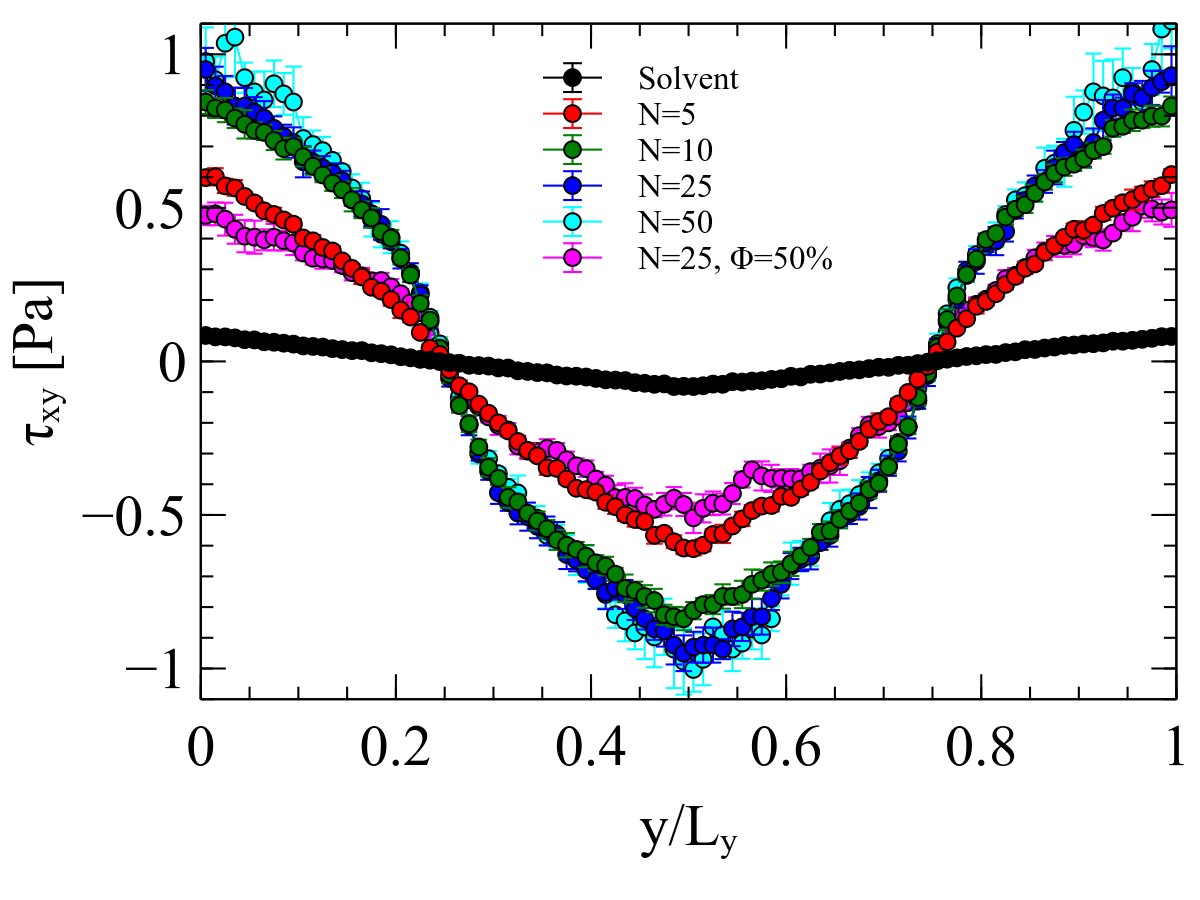
Attached is an image with the stress output for a system of 31k particles, including:
Solvent only
Melts (5,10,25,50) particles per chain
Solution 25 particles per chain at 50%
All the stress profiles should agree with each other and with the imposed one tau_xy= rhogxLy/4*(1-4*y/Ly). Since the simulation seems to be working properly (i.e. velocity profiles are correct), I am assuming there is something wrong with the stress calculation.
#Additional settings
bond_style fene # or lj 1 1 1
bond_coeff 1 {H} {r0} 0 0
special_bonds fene
mass 1 {sdpd_mass}
set group all meso/rho {sdpd_rho}
pair_style hybrid/overlay sph/rhosum 1 sdpd/taitwater/isothermal {sdpd_temp} {sdpd_eta} 28619
pair_coeff * * sph/rhosum {h}
pair_coeff * * sdpd/taitwater/isothermal {sdpd_rho} {sdpd_c} {h}
fix integrate_fix_full all meso
Any advice on what I could be missing is welcomed!
Thanks,
David