Dear LAMMPS Users,
I am a new user of LAMMPS. I am working on the simulation of Ag nanoparticle (100 Ag atoms) in alcoholic solution using EAM method for Ag and lj/cut for alcohol molecules . Since, I would like to simulate charged (for example, Ag5+, Ag10+ ) systems, therefore I use coul/wolf with damping parameter to account columbic interactions (pair_style hybrid/overlay lj/cut 25.0 coul/wolf 0.135 15.0 eam). The system charges are equilibrated using QEq/point charge model (chi and eta parameters are taken from Rappe and Goddard paper). According to QEq, the electronegativites are equilibrated and resulting total charge of the system is zero. However, I would like to keep the entire system as charged one, for example, If I mention the net charge on Ag particles as +5, the overall net charge must be preserved during the simulation.
My question is, Does anyone know how to fix (keep) the net charge of the system in this case?
Thanks in advance.
Regards,
Veerapandian
Dear LAMMPS Users,
I am a new user of LAMMPS. I am working on the simulation of Ag
nanoparticle (100 Ag atoms) in alcoholic solution using EAM method for Ag
and lj/cut for alcohol molecules . Since, I would like to simulate charged
(for example, Ag5+, Ag10+ ) systems, therefore I use coul/wolf with
damping parameter to account columbic interactions (pair_style
hybrid/overlay lj/cut 25.0 coul/wolf 0.135 15.0 eam). The system charges
are equilibrated using QEq/point charge model (chi and eta parameters are
taken from Rappe and Goddard paper). According to QEq, the
electronegativites are equilibrated and resulting total charge of the
system is zero.
This is not correct. According to QEq, total charge of the system is a
*constant*. Therefore your system net charge can be any value, which is a
value that will remain the same throughout your simulation. However, it
has been seen that the matrix inversion (used by qeq/point) method could
result in numerical instability when the system net charge is not zero.
This is the reason qeq/point enforces global charge neutrality. Damped
dynamics (used by qeq/dynamic), on the other hand, does not seem to have
this problem, so it only requires charge conservation not neutrality.
In simple words, you can switch to qeq/dynamic and that would be what you
are looking for.
Ray
Another note, coul/wolf is an effectively short-range, truncated summation method. If you don’t have global charge neutrality, you may observe a large energy fluctuation depending on the chosen alpha factor (0.135 in your case). Careful tests and analysis are recommended.
Ray
Dear Ray,
Sorry for the late reply.
Thank you very much for your explanation and suggestion. I will use qeq/dynamic instead of qeq/point in my case.
I have come across this following thread, explaining that qeq/dynamic total energy drift is worse than other methods (qeq/point, no qeq). Does qeq/dynamic method consistent in my case?
http://lammps.sandia.gov/threads/msg49152.html
Another question is, I am using EAM+coul/wolf (interaction between charged atoms) with qeq/dynamic charge equilibration for metal atoms, Does the electrostatic energy term include the energy response from E_eam along with coulomb term? for example: E_electrostatic = E_coul + E_eam, because some metal atoms are charged which I would also take into account.
Thank you
Veerapandian
EAM is not affected by charge on the atoms since it has a charge-independent functional form. Your electrostatic term will only come from Coulomb, which changes with changing charge values.
Ray
Dear Ray,
Thank you for your reply. I have a question about computing charges using qeq/dynamic with two different potentials (EAM and lj/cut for Ag metal).
I perform charge equilibration in two different cases
-
EAM/Alloy potential for Ag with qeq/dynamic (Ag atoms only)
-
lj/cut potential for Ag with qeq/dynamic (Ag atoms only)
If I understood correctly, charge equilibration has been performed in the system which is independent to the potential that I use for Ag atoms (EAM and lj/cut in my case). After simulation, I compared the charges on Ag atoms, I got different charges (I have attached the plot for your reference). I would expect that the charges should have the same values? Could you please tell me why I received different charges? Your suggestion will be highly appreciated.
Thanks in advance.
veera
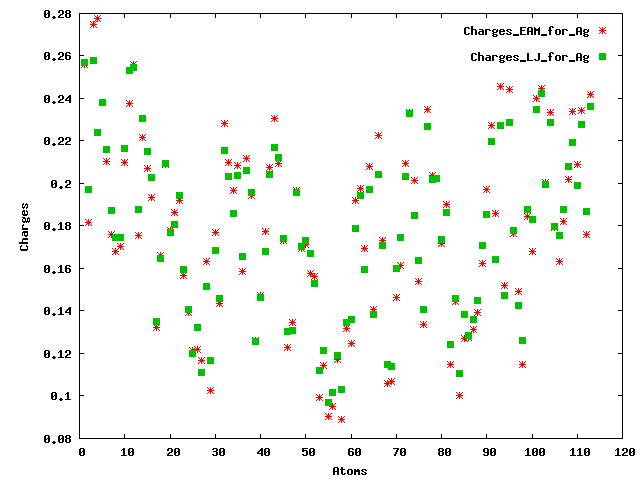
On 0th step, yes, charges would be independent of pair_styles used, but as soon as you run dynamics, even for just 1 step, atom positions can change, which can affect atom charges - this is likely what you are seeing.
Ray
Thanks for your reply with nice explanation. Now It’s clear to me.
Veera