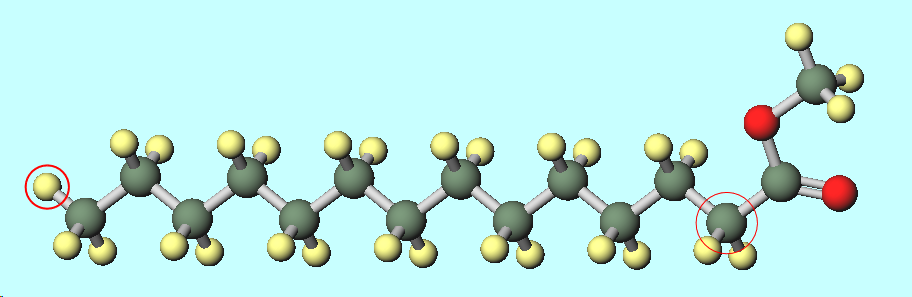
Thankyou for your kind suggestions the best I have found in months. I have fixed a few errors in my program and found the appropriate file (attached here)generated by the Topotools in TK Console. I am still struggling to find the appropriate potential for the methylmyristate compound I am using. I am planning to use the LJ/cut potential only because I found a tutorial for a octane molecule using the same potential. I plan to use the pair_coeff of CH3 and CH2 molecules from the tutorial but :
it is not that simple. atom types in classical force field are chosen by “chemical nature”.
1- I am not sure how to find the pair_coeff for the other two species C and O.
please read up on how to assign atom types and how (classical) force fields function in a (good) text book on MD simulations like “Understanding Molecular Simulations” by Frenkel/Smit. also discuss with your adviser. what you are asking are not LAMMPS questions, but general MD questions, which are outside the scope of this mailing list.
2- Please also advise me if I have grouped them correctly in CH3, CH2, C and O.
that depends on the force field chose. same as above, this is not a LAMMPS question, but a manifestation, that you are lacking proper training. that is something that your adviser/supervisor/tutor has to teach you.
3- Please direct me towards some literature where I can understand how these pair coefficients are derived.
again, this is a question outside the scope of the this mailing list. please discuss with your adviser, read up on this in a proper text book and then research which force field is best applicable to the compound and thermodynamic state of the system you want to investigate and then look up the publication describing that force field. popular choices are CHARMM, Amber, OPLS/AA, GROMOS, each have different strategies and different mechanisms/rules how to assign atom types (and thus non-bonded and bonded parameters) and how to assign (partial) charges to model the charge distribution of your compound. the easiest choice in your case is possibly OPLS/AA, which uses an increment system to determine the (partial) charges.
however, as mentioned before, this is a topic for a discussion with your adviser/supervisor or senior/experienced colleagues, not this mailing list.
axel.