Perhaps Aidan wants to comment. But I would start
with a simpler test than maintaining an equilibrium
structure during a run. Can you reproduce energy
and forces from the paper for a few snapshots of
simple geometries?
I agree with Steve. Before doing a simulation, calculate simple properties
such as defect energies, elastic constants, and compare with the paper.
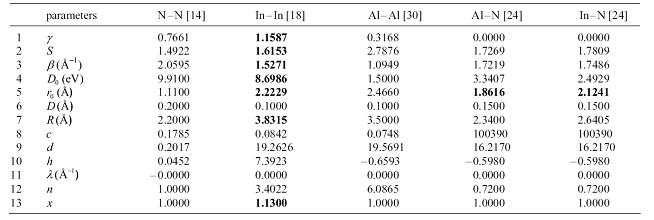
Also, using the authors' descriptions, carefully calculate all parameter
values for cross-interactions. Finally, read the LAMMPS documentation to
make sure you understand how LAMMPS defines the input parameters.
The tersoff doc page has great details about the format of the potential
file, and there are several examples. There’s not really more I can do for you.