Dear colleagues
I’m using Lammps 7 Aug 2019 version.
I am trying to carry out polymer chain simulations, but all simulations have crashed.
An example of an error I’m getting is as follows:
ERROR on proc 8: Bond atoms 3571 3573 missing on proc 8 at step 3731172 (…/ntopo_bond_all.cpp:63)
Looking at previous answers on the Lammps forum, I noticed that this kind of error is due to bad dynamics. I checked the points below (solutions from previous answers) and I still haven’t been very successful:
- I used the command “neigh_modify delay 0 every 1 check yes”;
- I checked the force field parameters (maybe the published ones are wrong);
- I reduced the timestep.
Now I am looking at the issue of initial geometry, which I think should be the (real) problem.
First, I build a polymeric chain in a given software:
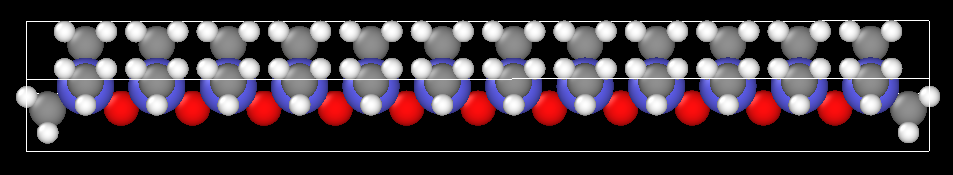
Later, I built my system with “n” polymer chains (i.e., my initial geometry used in Lammps):
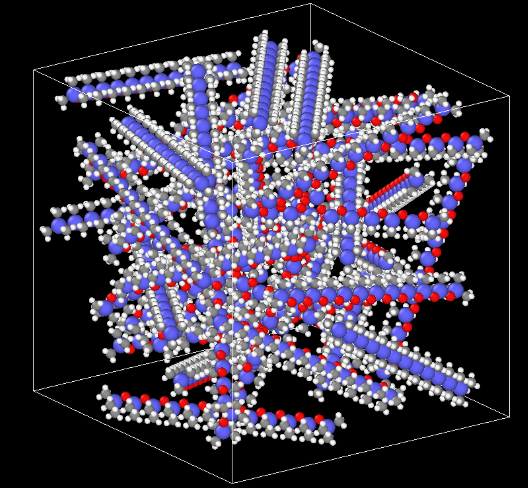
From the second figure (e.g., my initial geometry), I perform a minimization step, 2 ns of equilibration nvt, 3 ns of equilibration npt and 5 ns of production npt. Example below:

My question is, the most correct thing would be to minimize the energy of a single polymeric chain, apply a equilibration step (Langevin dynamics at 5000 K as in the example of the Lammps tutorials https://icme.hpc.msstate.edu/mediawiki/index. php / LAMMPS_Polymer.html) and only then, build my system with “n” polymeric chains?
Sorry if my question was too out of scope for the mailing list.
Any suggestion is appreciated.
Thanks in advance for your time.
Best regards,
Emerson Parazzi Lyra
PhD candidate
School of Chemical Engineering
University of Campinas
500 Albert Einstein Ave, Campinas, SP, 13083-852, Brazil
Email: [email protected]
Orcid ID: https://orcid.org/0000-0002-7969-3764