Dear colleagues,
I am trying to understand how the intermolecular potential is calculated by the Lammps to obtain intermolecular potential parameters properly from quantum mechanics calculations (to use them later in Lammps simulations, i mean).
I intend to use pair_style lj/cut/coul/long, whose equations are described below:
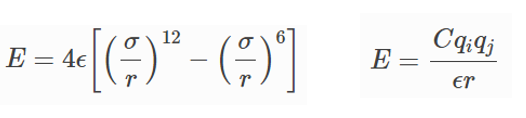
My question is: what would be the distances between atoms (i.e., “r”) to be considered in Lammps intermolecular potential energy calculations (i.e., both Lennard-Jones and Coulomb potentials)?
For example, in the pair of methane molecules below (where C is the carbon, H is the hydrogen and numbers are IDs):
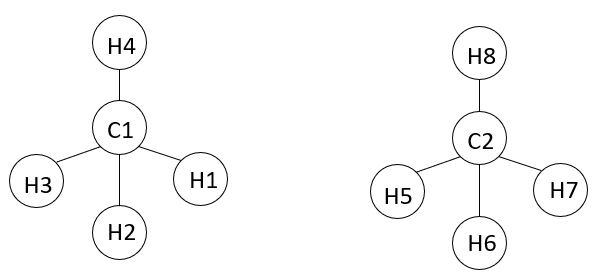
1 - Would Lammps consider “ij” interactions of only different molecules when calculating the intermolecular potential (i.e. Lennard-Jones and Coulomb)? (e.g., C1C2; C1H5; C1H6; C1H7; C1H8; C2H1; C2H2; C2H3; C2H4; H1H5; H1H6; H1H7; H1H8; H2H5; H2H6; H2H7; H2H8; H3H5; H3H4; H3H7; H4H8).
2 - Or would Lammps consider all possible “ij” interactions? (e.g., C1C2; C1H1; C1H2; C1H3; C1H4; C1H5; C1H6; C1H7; C1H8; C2H1; C2H2; C2H3; C2H4; C2H5; C2H6; C2H7; C2H8; H2H1; H3H1; H4H1; H5H1; H6H1; H7H1; H8H1; H3H2; H4H2; H5H2; H6H2; H7H2; H8H2; H4H3; H5H3; H6H3; H7H3; H8H3; H5H4; H6H4; H7H4; H8H4; H6H5; H7H5; H8H5; H7H6; H8H6; H8H7).
If “1” is true, for a situation where I have 5 atoms connected in sequence in the same molecule (e.g. 1-2-3-4-5), would I have to compute the interactions of atoms 1-5 in the same molecule?
Sorry in advance if my question was too “basic” or if it is out of the scope of this mailing list.
Thanks for a while.
Best regards,
Emerson Parazzi Lyra
PhD candidate
School of Chemical Engineering
University of Campinas
500 Albert Einstein Ave, Campinas, SP, 13083-852, Brazil
Email: [email protected]
Orcid ID: https://orcid.org/0000-0002-7969-3764