Hi all,
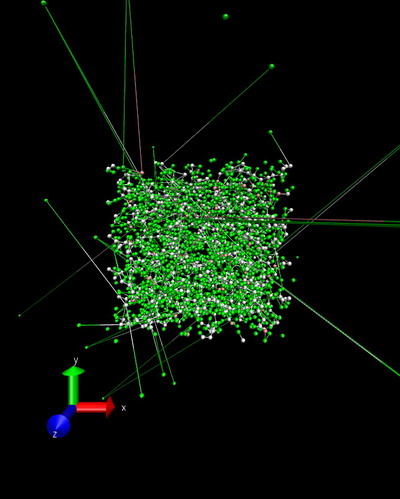
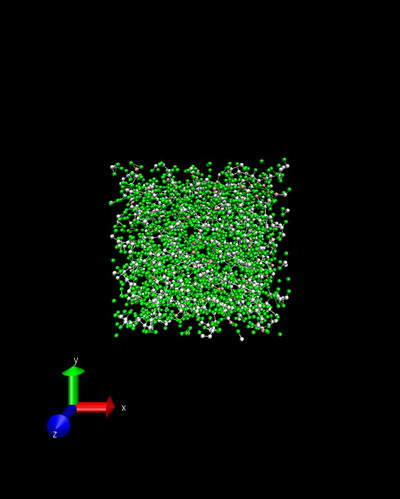
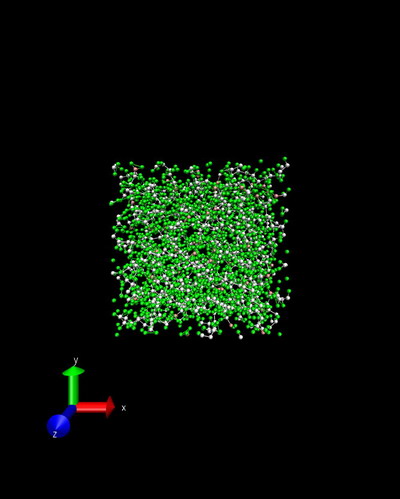
I’m trying to simulate polyethylene using airebo forcefield. The input details can be found in attached output log.lammps. During first energy minimization the structure is stable, as shown in initial.jpg and minimize.jpg. However, the temperature increases to an unreality value and error of lost atoms commences during initial stage of following npt relaxation. The snapshot of npt.jpg shows there are extremely long bonds within the structure. I also try nvt instead of npt, but the same problem remians. I have no idea about the reason beyond this phenomena, could you give me some advice? Any suggestion is highly apprecaited!
P.S. Since there is size limitation of attachment, here only part of the corresponding data file is presented for reference.
LAMMPS 2005 data file for Polyethylene
3100 atoms
3050 bonds
6000 angles
8550 dihedrals
0 impropers
3 atom types
4 bond types
7 angle types
7 dihedral types
0.000000000 28.620000000 xlo xhi
0.000000000 28.620000000 ylo yhi
0.000000000 28.631400000 zlo zhi
Atoms
1 0 1 0.100000 4.204052588 7.377315847 3.931162030
2 0 2 -0.300000 4.860980653 7.796637263 4.689492951
…
Bonds
1 1 1 2
2 2 2 3
…
Angles
1 1 1 2 3
2 2 1 2 4
…
Dihedrals
1 1 1 2 3 6
2 1 1 2 3 7
…
Best,
Yusong He
log.lammps (10.1 KB)