Hi LAMMPS users,
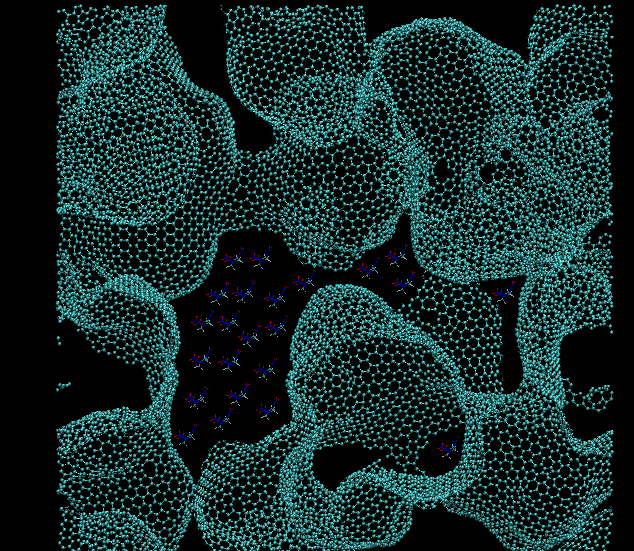
I am trying to create an initial configuration involving a graphene foam
and then fill the voids of the foam with another compound. Ideally it
should look something like (ref: attached image: 'initial_system'). I have
tried the following:
** Used VMD to manually drag and drop the molecules in the void. But this
is difficult as the image breaks up if zoomed too much to make the
placement exact. I was able to make an initial configuration but the LAMMPS
simulation gave the following error.
*WARNING: Fix qeq/reax CG convergence failed after 200 iterations at 1 step
(src/src/USER-REAXC/fix_qeq_reax.cpp:739)*
*ERROR on proc 29: Non-numeric positions - simulation unstable
(src/src/nbin.cpp:125) *
Have also tried to work around this by running it at a very low temperature
of 0.01 K and also changing the convergence criteria and making it a bit
more tolerant. But neither of that worked.
The input script for this has been attached with this email.
I am looking an effective way to fill the pores. Things which might help:
1. Any post-processing tool/code that helps to extract the location and
size of the pores in graphene foam so that targeted placement of molecules
can be done.
2. A better visual tool - something similar to VMD, which can help with
manual drag + drop and placement.
Any help is appreciated, and thank you in advance.
Regards,
Suman
in.RDX (1.5 KB)
You could just create/load the two subsystems on top of each other and then use some smart scripting to detect overlapping atoms (aka close contacts) and then delete the entire molecule.
This can be done with both LAMMPS and VMD, but requires that you have bonds, so they can determine what constitutes a “molecule” for removal, which would have to be removed afterwards, since reaxff does not use explicit bonds.
Hi Axel,
Thank you for the suggestion. I am in the process of writing a script to detect the overlapping atoms and delete them. There is another approach I was attempting. The RDX molecule I am planning to place in the voids of graphene foam has an approximate size of 5x5x5 [units in Angstrom]. Now I wrote a script to find how many empty spaces of size 5x5x5 are present in the foam. This way I can know the pore size distribution and do a targeted placement of the molecules in the voids of the foam. I am attaching my input, molecule and data files. I am currently facing an error of:
ERROR on proc 0: Molecule auto special bond generation overflow (src/src/molecule.cpp:1200)
I did find another post on LAMMPS with the same error, but not able to connect the dots.
Am I missing something simple and obvious here?
yes. your added molecules require more storage for excluded neighbors and bonded interactions than the graphene foam does.
however, those numbers are locked in when the box is created. using a data file does that implicitly and the numbers are chosen to exactly fit the needs of the system in the data file.
if you then later add molecules (either from a data file or through the molecule command) you cannot expand this. instead you have to use the various “extra” keywords with the first read_data command to reserve additional space. please see the manual for a more detailed explanation.: https://docs.lammps.org/read_data.html