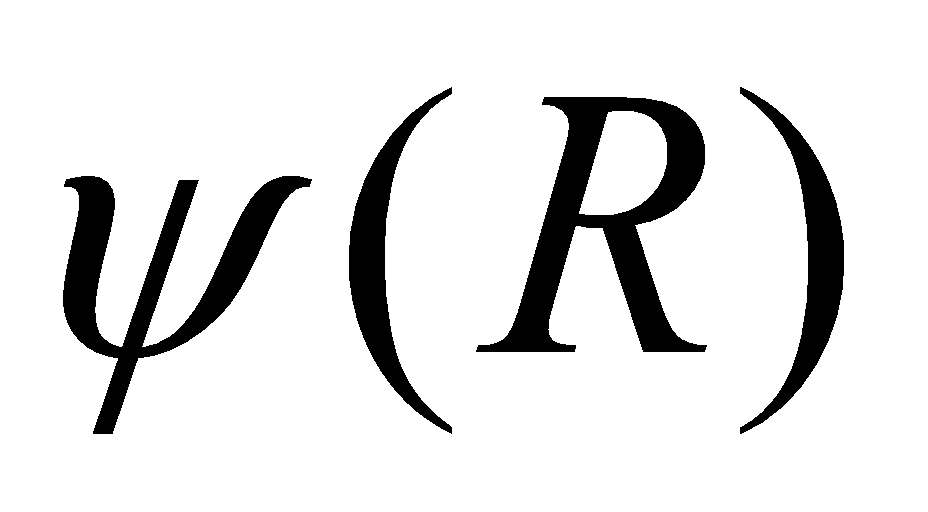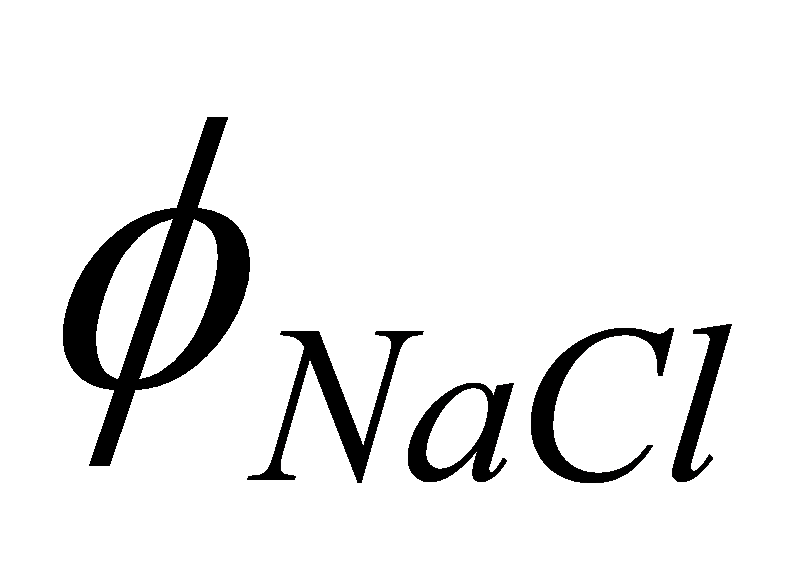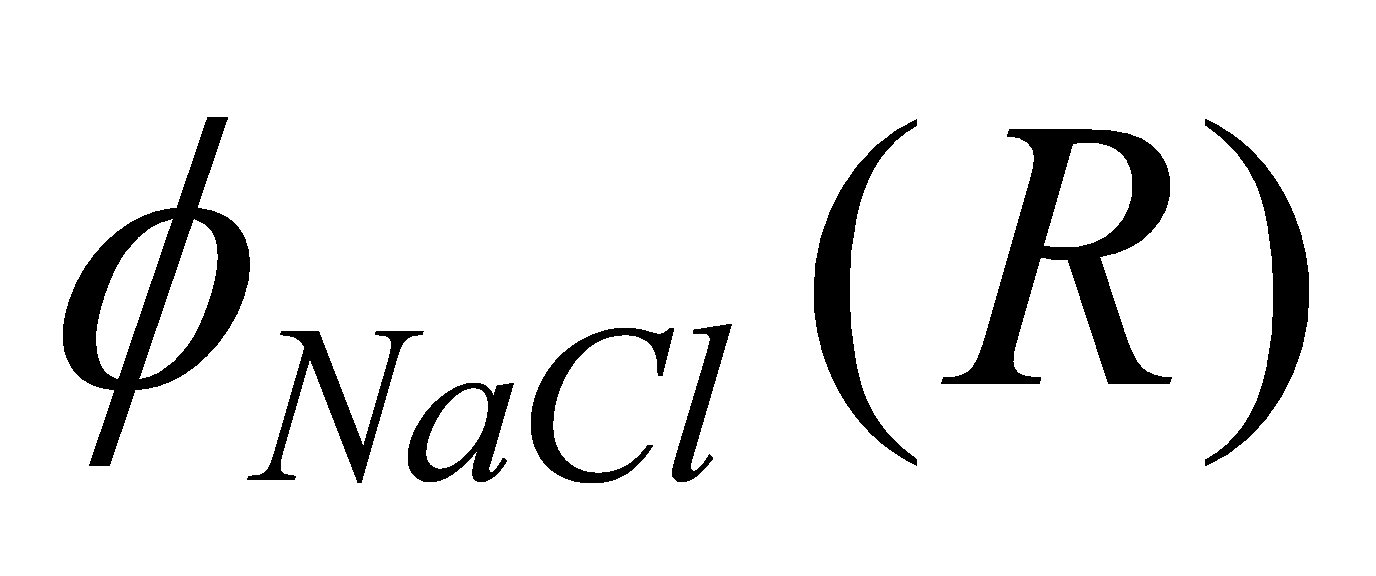I have a few questions regarding the 2NN meam treatment in MEAM code:
i) In subroutine get_densref, only rho0 (and not, rho1, rho2, and rho3) is modified for 2NN treatment. Is it simply because the 2NN implementation is currently limited to fcc or bcc lattices where shape functions are all zero? That is, in order to apply 2NN treatment to other structures should all partial densities need to be modified in accordance with the specific structure?
ii) In subroutine compute_pair_meam, it is not clear to me why a value of 10 has been chosen for nmax. >From Lee and Baskes (PRB, 62, 2000, pp. 8564) paper, it seems that nmax is arbitrarily chosen to fit experimental value of energy. However, it makes more sense to me from the formulation developed in the same paper, that nmax = 1, as has been done for the b1 structure in the code. Am I right in thinking that nmax=1 the more ‘appropriate’ choice?
In question (ii) below, I was wrong: nmax =1 is not the 'appropriate'
choice. For b1 structure, nmax is probably taken as 1 because the ratio
of 2nd neighbor to 1st neighbor distance is very large (=sqrt (2)) which
would make much smaller than . Please comment if this is correct or
not.
Thanks
Rohit
Senior R&D Engineer
Hawthorne &York Intermational
4645 S 35th Street
Phoenix, AZ 85040
Ph. +1 602 275 4633, Ext 210
Fax: +1 602 273 4503
As far as I know, the current MEAM lib does not support many structures with 2nn option since it is very challenging to generalize the formalism. Probably the developer does not have time and efforts to do that.
The nmax is always larger than or equal to 10. IF you look at Lee’s later paper, it was stated that the comutation procedure will repeat until overall agreements between calculation and the target property value (experimental or ab initio) are obtained. In his own code, he also used sth like " nmax = 12 ". I dont know if your explanation regarding the b1 structure is correct or not but the ratio of fcc is also sqrt(2).
Thanks for your response. It does seem that the 2NN meam portion is not
well-developed. I am trying to implement the 2NN potential for a
particular structure. The average weighting factors t11av, t21av,
..t31av, t32av, have not been modified by for the 2NN analysis. Is this
okay or should it ideally be modified appropriately?
Thanks
Rohit
PS: My explanation for b1 structure was wrong. The real reason perhaps
is that because of the type of neighbors in b1 structure (all 1NN are
opposite type and all 2NN are of the same type), when the equation for
energy using 2NN is formulated in the pattern of 1NN, we get the
equation:
Since is calculated only at R, i.e. there is only 1 term, to
calculate we don't need to iterate if we use the potentials for
Na-Na, Cl-Cl interactions based on 1NN analysis here.
(i) Where can I find this latest version? I downloaded the 15th Jan
version today from Sourceforge.net and it has the t11av values.
(ii) If I follow the formulation given in Chapter 4 of 'Numerical Tools
for Atomistic Simulations' by Gullett, Wagner and Slepoy, it would make
sense to use the average values t11av, t21av, and t31, for Gamma and
elemental values t11, t21, and t31, for Gamma(ref) (since the average
values become the elemental values for the reference structure for the
element 1). Is that what is done in the new version you mentioned, or
has t11av never been used? That is, has the Eq. 4.9 in the above source
never been used in the new version?
(iii) Does anybody know of any source that describes the calculation of
shape factors? From the values given for fcc, bcc, hcp and diamond cubic
in Baskes PRB, vol 46, 1992, pp 2727, I figured that the shape factors
are summations similar to partial electron density terms but without the
atomic electron densities.
Re (i) and (ii) below: A recent (mid-2009) change to the MEAM formulation changed the way that Gamma_ref gets computed; as Rohit points out, it now uses the elemental values of t to compute Gamma_ref (eqn. 4.6 in the Gullet et al. report), and the average values (eqn. 4.9) to compute Gamma itself (eqn. 4.4). This is not quite consistent with what’s written in the Gullet report, which uses average values for both quantities. The change was made after some discussions with Mike Baskes on what is the correct thing to do. Mike made similar changes in his own MEAM code, dynamo.
Re (iii): The shape factors are the coefficients of t1, t2, and t3 in the expression for Gamma that are computed in the reference structure. They can be computed by assuming only nearest-neighbor interactions. These are zero because of symmetry for many structures, like fcc – in those cases, cancelations in the summation over neighbors lead to zero values.
Re the 2NN formulation: It’s true that this is not well developed for all cases, largely because it’s hard to write out a formulation for the general case. Certain structures are straightforward, and many of those are captured in the code, but if one you’re interested in is missing you can probably do so without much change to the code (provided that case can be formulated at all). As for the number of terms used in the summation (nmax=10), I think I copied this from Mike Baskes’ original code; the convergence should be very good with 10 iterations (those extra terms die away quickly), but feel free to play with increasing this.
Thanks for your reply most of which confirms what I thought.
(i) If we simply consider that Gamma_ref is the same as Gamma except
that Gamma is calculated based on the actual environment of an atom of
element i in the alloy reference structure whereas Gamm_ref is
calculated for the reference structure of element i, then by analogy
s(k)* rho_io^2 is the rhobar(k)^2 term, whereas (Z_io^2)* (rho_io^2) is
the rhobar(0)^2 term; rho_io^2 term cancels out in division. ti(k)'s
then become the values for elemental reference structure when
calculating Gamma_ref, and the values for alloy structure when
calculating Gamma. This interpretation makes what is written in the
Gullet et al. report consistent with what you said is currently being
done in the MEAM codes. I've calculated shape factors based on this
interpretation, and it comes out to be consistent with that reported in
Baskes paper for fcc, bcc, dc, hcp structures.
Could you please comment if the above is the right interpretation of
shape factors? It may have some bearing on specific situations.
(ii) Where may I find this new version of MEAM formulation? The latest
versions from www.sourceforge.net <http://www.sourceforge.net/> and http://lammps.sandia.gov/> still have the
older scheme where alloy averaged t1, t2 and t3 are used. I have already
made these changes, but still want to look if there is anything else
that is different from the version that I have.
Thanks
Rohit
Senior R&D Engineer
Hawthorne &York Intermational
4645 S 35th Street
Phoenix, AZ 85040
Ph. +1 602 275 4633, Ext 210
Fax: +1 602 273 4503
(ii) Where may I find this new version of MEAM formulation? The latest versions from www.sourceforge.net and http://lammps.sandia.gov still have the older scheme where alloy averaged t1, t2 and t3 are used. I have already made these changes, but still want to look if there is anything else that is different from the version that I have.
What is currently on SF, is the 15Jan10 version. There have been no changes to MEAM
since then, so it is current.