Dear all,
I used 23Sept-2010 version of lammps on two intel clusters.
The code was compiled with openmpi, see Makefile in attachment.
Si-C - interactions are described by MEAM potentials.
Through the execution of NVE calculation in SiC system with two
spherical voids temperature and total energy of the system increase.
In the case of ideal SiC the temperature stays at 1000 K as it's suppose to be.
For the Si with voids temperature behavior is normal.
The earlier version of lammps (7Jul09) does not have this problem.
Does anybody have this problem?
Sincerely,
German Samolyuk.
There was another email in the last couple weeks of someone
saying MEAM a year+ ago did something "right" and the current
one is "wrong". There have been changes to MEAM in that timeframe
but they were to make the potential more up-to-date with the most
current MEAM in the literature. So right/wrong is open to some
interpretation. I'm CCing Greg Wagner on this - he may want
to look into this. Greg - rising temperature sounds ominous, like
possibly a bug - or maybe the newer MEAM requires a smaller
timestep?
Thanks Steve,
I'd like to mention that for perfect SiC everything is OK. The problem
rises in the system with voids.
For Si with voids everything is OK too.
German
Steve,
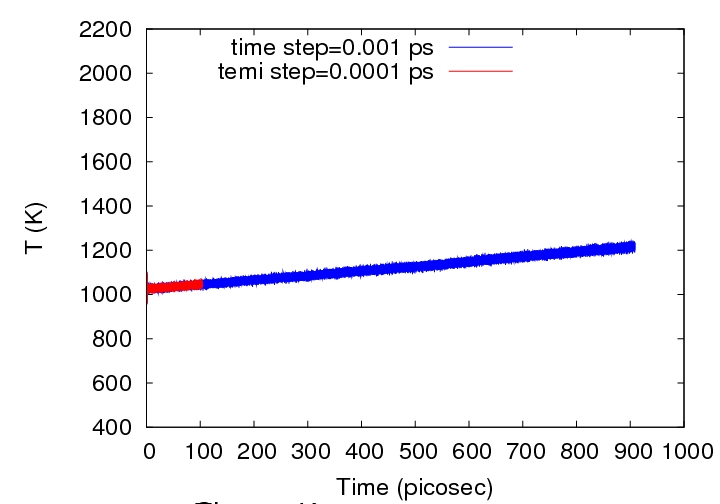
Attached please find jpg figure with temperature dependence of time.
red color corresponds to time step 0.001 ps and blue one - 0.0001 ps.
I also compared output from version lammps-7Jul09 , the last version
correctly working with SiC with bubble I have,
and the lammps-23Sep10.
The difference in temperature, pressure, energy ... starts at
iteration number 17 and rise from difference in forces action to
atoms:
#ITEM: ATOMS id x y z fx fy fz type
1063 7.84478 14.3565 12.1216 -4.76575 1.67113 1.00169 2
1063 7.84478 14.3565 12.1216 -4.69355 1.52205 0.839464 2