Hi all,
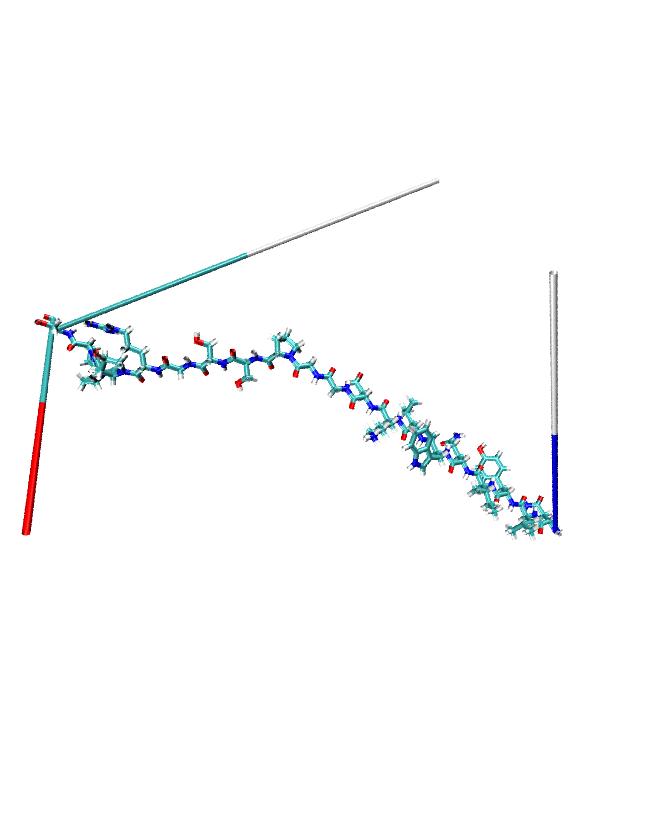
I used lammps to minimize a small peptide. When I loaded the dump file into vmd. some atom positions seem wrong to me as in the attached structure. Can somebody tell me what is wrong?
Thanks a lot.
Jianping
Hi all,
I used lammps to minimize a small peptide. When I loaded the dump file into vmd. some atom positions seem wrong to me as in the attached structure. Can somebody tell me what is wrong?
Thanks a lot.
Jianping
Hi all,
I used lammps to minimize a small peptide. When I loaded the dump file into
vmd. some atom positions seem wrong to me as in the attached structure. Can
somebody tell me what is wrong?
jianping,
this is very difficult to say without knowing exactly
how and in which format you created the trajectory.
also you seem to use the perspective projection.
my guess is that you have atoms going across box boundaries.
cheers,
axel.
HI Axel,
Thanks. The attachments are my data and input file for the minimization. I am new in lammps. Can you tell me what is wrong?
Jianping
mytrp-cage.in (896 Bytes)
mytrp-cage.data (167 KB)
HI Axel,
Thanks. The attachments are my data and input file for the minimization. I
hi jianping,
am new in lammps. Can you tell me what is wrong?
you input file doesn't even run!!
how is it possible that this was working for you?
but simply from looking at the data file it seems
obvious that the box is determined by the min-max
of the coordinates. since you have no solvating
water molecules, those are the boundaries of the
peptide and thus what you are seeing are atoms
that get wrapped across the periodic boundaries.
since you furthermore request the resulting coordinates
to be stored in fractions of the system cell and
the origin of your cell is not at 0.0,0.0,0.0,
you already have some wrapped coordinates in the
very first frame.
the lammps molfile plugin doesn't handle the image
counter from the lammps dump file format currently.
in any case, you have to enlarge the simulation cell
for the calculation to have any meaning at all.
check out the attached image that shows how close
the various periodic images are. for an isolated
molecule in gas phase actually using no PBC would
be more adequate then the you don't need long-range
coulomb with pppm or ewald either.
cheers,
axel.
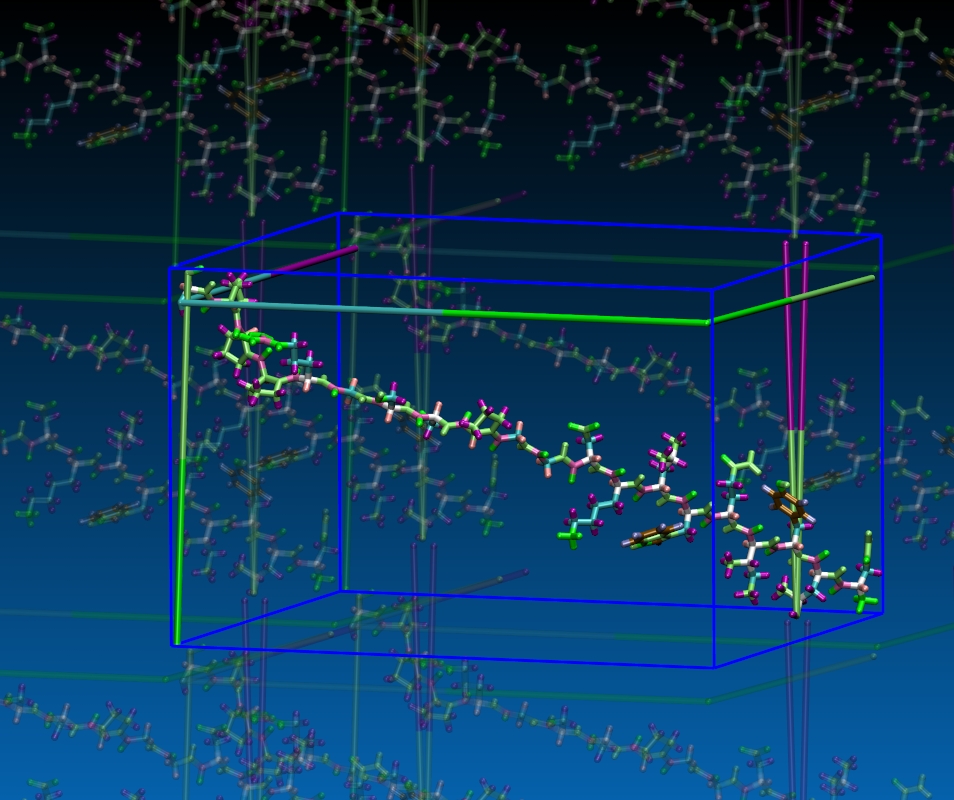
Thanks Axel. when I enlarge the cell in the data file, it is working. But when I simply common out the box in the data file, it doesn’t work. How can I get rid of the PBC from the data file and input file? Also, I like the beautiful image you sent. Can you tell which software you used to generate it from the data file?
Jianping
When you dump atom coords for a molecule, if you want
them unwrapped so they don't straddle the periodic boundary,
then look at the dump custom command and some of its options
like xu,yu,zu. This may help with the viz.
Steve
jianping,
Thanks Axel. when I enlarge the cell in the data file, it is working. But
when I simply common out the box in the data file, it doesn't work. How can
please have a closer look at the documentation. you _always_ need to
provide a simulation cell. if for nothing else, LAMMPS needs that
information for parallelization.
I get rid of the PBC from the data file and input file? Also, I like the
check out the boundary command. just enlarge the box by a bit
and then use e.g., boundary s s s
beautiful image you sent. Can you tell which software you used to generate
it from the data file?
the image was done with VMD.
axel.
When you dump atom coords for a molecule, if you want
them unwrapped so they don't straddle the periodic boundary,
then look at the dump custom command and some of its options
like xu,yu,zu. This may help with the viz.
yep. but most likely not for VMD. the lammps plugin
in VMD currently only handles text mode 'atom' style dumps.
to get an unwrapped trajectory while using periodic boundaries
which is readable by VMD one either can dump into .dcd or .xtc
format with 'dump_modify <dumpid> unwrap yes' or just use the
join and unwrap functions of the pbcwrap plugin bundled with VMD
to try and reconstruct an unwrapped trajectory. the latter doesn't
always work without some tinkering, though, and requires the latest
test version of VMD.
cheers,
axel.
Thanks a lot.
Jianping