Dear Lammps users,
Is it possible to provide parallel and perpendicular orientation for ellipsoidal particles using lammps?
How can I do?
Can the order/orientation parameter be measured?
Thanks in advance
Dear Lammps users,
Is it possible to provide parallel and perpendicular orientation for ellipsoidal particles using lammps?
How can I do?
Can the order/orientation parameter be measured?
Thanks in advance
Not sure what you are asking. If you mean, can you initialize
the orientation of one or more ellipsoidal particles, then yes.
You can list them in the data file with the orientation
you want (as quaternions). Or you can use the "set quat"
command to rotate them to new orientations.
Steve
2009/10/1 ahmet yıldırım <[email protected]...>:
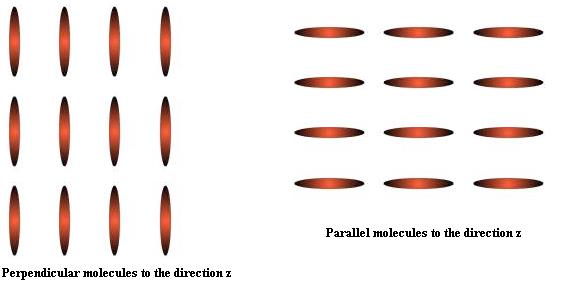
I want to simulate the oriented ellipsoidal particles ( all particles in simulation box).
set group all quat 0 0 1 0 #theta, a,b,c
can this command do its? For example: How can I created perpendicular molecules to the direction z?
For example: attached figure
Thanks in advance
02 Ekim 2009 18:41 tarihinde Steve Plimpton <[email protected]> yazdı:
If you read the doc page on the set quat command
it explains what its orientation arguments mean.
So I would try it out and visualize the results, until
you get what you want.
Steve
2009/10/13 ahmet yıldırım <[email protected]...>:
This command "set group all quat 0 0 1 0 " provide the orientation parallel to the direction of z but it is doing for first step.(I see using visualize).How can I apply/perform to all steps it?
Furthermore, I removed this command" set group all quat/random 18238".Does it make sense to remove it?
Thanks in advance
input file:
units lj
atom_style ellipsoid
dimension 3
boundary p p p
lattice sc 0.01
region box block 0 8 0 8 0 16
create_box 1 box
create_atoms 1 box
set group all quat 0 0 1 0
mass 1 1.0
shape 1 1 1 3
.
.
.
14 Ekim 2009 16:54 tarihinde Steve Plimpton <[email protected]> yazdı:
This command "set group all quat 0 0 1 0 " provide the orientation parallel to the direction of z but it is doing for first step.(I see using visualize).How can I apply/perform to all steps it?
Furthermore, I removed this command" set group all quat/random 18238".Does it make sense to remove it?
Thanks in advance
input file:
units lj
atom_style ellipsoid
dimension 3
boundary p p p
lattice sc 0.01
region box block 0 8 0 8 0 16
create_box 1 box
create_atoms 1 box
set group all quat 0 0 1 0
mass 1 1.0
shape 1 1 1 3
.
.
.
15 Ekim 2009 00:11 tarihinde ahmet yıldırım <ahmedo047@…24…> yazdı:
I don't understand what you're asking. Indeed, the set command
will set the orientation once. I assume you want the ellipsoids
to change their orientation during the simulation after that. If you
really want to freeze them at their initial orientation, then don't integrate
them via the fix nve/asphere command.
Steve
2009/10/14 ahmet yıldırım <[email protected]...>:
Thanks for your response.
Yes.I want the ellipsoids to change their orientation during the simulation.That is, I want to calculate thermal conductivity(using muller-plathe method) of the oriented ellipsoidal particles.
This is not possible according to you’ve said.Alright,Do you have a recommendation to me?
Thanks in advance
16 Ekim 2009 16:42 tarihinde Steve Plimpton <[email protected]> yazdı:
If you just do an initial "set", then use fix nve/asphere, then
the ellipsoidal particles will reorient during the simulation. I don't
know what that means for thermal conductivity. The MP method
will only change their translational energy, not their orientational
motion.
Steve
2009/10/17 ahmet yıldırım <[email protected]...>:
I think I could not tell exactly my problem. if possible, can you look at attached file?
Thanks in advance
20 Ekim 2009 02:34 tarihinde Steve Plimpton <[email protected]> yazdı:
abc.doc (28.5 KB)
Dear Lammps users,
This command "set group all quat 0 0 1 0 " provide the parallel orientation to the direction of z
but the orientation is changing after a few steps.I want to freeze them at their initial orientation(0 0 1 0).How can I apply to all steps it?Is this possible?
Thanks in advance
20 Ekim 2009 22:07 tarihinde ahmet yıldırım <ahmedo047@…24…> yazdı:
As I've said before, if you don't want the particles
to move or re-orient during a dynamics run, then
don't time integrate them. I.e. don't put them
in a group that is assigned to fix nve or fix nve/asphere
Steve
2009/10/26 ahmet yıldırım <[email protected]...>:
Dear Steve,
I am only using npt/asphere but the orientation of particles is still changing.Whereas I want to freeze them at their initial orientation(0 0 1 0). I want particles to move during a dynamics run but I do not want to change the orientation of particles.
Thanks
My input script:
units lj
atom_style ellipsoid
lattice sc 0.01
region box block 0 8 0 8 0 16
create_box 1 box
create_atoms 1 box
set group all quat 0 0 1 0
mass 1 1.0
shape 1 1 1 3
compute rot all temp/asphere
group spheroid type 1
variable dof equal count(spheroid)+2
compute_modify rot extra ${dof}
velocity all create 3.0 87287 loop geom
pair_style gayberne 1.0 3.0 1.0 4.0
pair_coeff 1 1 1.0 1.0 1.0 1.0 0.2 1.0 1.0 0.2
neighbor 0.8 bin
timestep 0.002
thermo_style custom step temp etotal vol epair press pe
thermo 100
fix 1 all npt/asphere 3.0 3.0 0.5 xyz 0.0 8.0 0.5
compute_modify 1_temp extra ${dof}
run 100000
unfix 1
fix 2 all npt/asphere 3.0 3.0 0.5 xyz 8.0 8.0 0.5
compute_modify 2_temp extra ${dof}
run 100000
26 Ekim 2009 17:40 tarihinde Steve Plimpton <[email protected]> yazdı:
Dear Ahmet,
I think I finally could understand what kind of system you want to simulate! but I think your system is not a real system! I mean you can not make such system in any laboratory and do physical experiments on it!! If I’m not right would you please explain in detail how could you create such system in a real lab and under exactly what physical condition this system should be kept? (perhaps using an external magnetic or electric field or so on.)
Zahra
If you want them to translate but not rotate, then
don't integrate the rotational degrees of freedom. I.e.
use fix nve, but not fix nve/asphere.
Steve
2009/10/26 ahmet yıldırım <[email protected]...>:
Dear Steve,
Firstly, Thanks for your helps. I did as you said.I used npt instead of npt/asphere.Eventually I obtained the parallel orientation to the direction of z. But I have two problem:
1.I entered Temperature=2.6 at initial configuration but temperature value is reaching approximately 1.7. That is, tempereture don’t reach to 2.6.
What should I do?
Should I remove two line the following in input file?
compute_modify 1_temp extra {dof}
compute_modify 2_temp extra {dof}
2.I don’t integrate the rotational degrees of freedom by using NPT as you said.I have obtained all the phases of ellipsodal particles by using NPT/asphere before.
can I obtained all the phases using NPT? Because In the isotropic phase the molecules are randomly aligned.
Thanks for everything.
Finally input File:
units lj
atom_style ellipsoid
lattice sc 0.01
region box block 0 8 0 8 0 16
create_box 1 box
create_atoms 1 box
set group all quat 0 0 1 0
mass 1 1.0
shape 1 1 1 3
compute rot all temp/asphere
group spheroid type 1
variable dof equal count(spheroid)+2
compute_modify rot extra ${dof}
velocity all create 2.6 87287 loop geom
pair_style gayberne 1.0 3.0 1.0 4.0
pair_coeff 1 1 1.0 1.0 1.0 1.0 0.2 1.0 1.0 0.2
neighbor 0.8 bin
timestep 0.002
thermo_style custom step temp etotal vol epair press pe
thermo 100
dump 1 all custom 100 dump.ellipse.gayberne &
id type x y z quatw quati quatj quatk
fix 1 all npt 2.6 2.6 0.5 xyz 0.0 8.0 0.5
compute_modify 1_temp extra ${dof}
run 100000
unfix 1
fix 2 all npt 2.6 2.6 0.5 xyz 8.0 8.0 0.5
compute_modify 2_temp extra ${dof}
run 100000
27 Ekim 2009 16:00 tarihinde Steve Plimpton <[email protected]> yazdı:
Dear Steve,
Firstly, Thanks for your helps. I did as you said.I used npt instead of npt/asphere.Eventually I obtained the parallel orientation to the direction of z. But I have two problem:
1.I entered Temperature=2.6 at initial configuration but temperature value is reaching approximately 1.7. That is, tempereture don’t reach to 2.6.
What should I do?
Should I remove two line the following in input file?
compute_modify 1_temp extra ${dof}
compute_modify 2_temp extra ${dof}
2.I don’t integrate the rotational degrees of freedom by using NPT as you said.I have obtained all the phases of ellipsodal particles by using NPT/asphere before.
can I obtained all the phases using NPT? Because In the isotropic phase the molecules are randomly aligned.
Thanks for everything.
Finally input File:
units lj
atom_style ellipsoid
lattice sc 0.01
region box block 0 8 0 8 0 16
create_box 1 box
create_atoms 1 box
set group all quat 0 0 1 0
mass 1 1.0
shape 1 1 1 3
compute rot all temp/asphere
group spheroid type 1
variable dof equal count(spheroid)+2
compute_modify rot extra ${dof}
velocity all create 2.6 87287 loop geom
pair_style gayberne 1.0 3.0 1.0 4.0
pair_coeff 1 1 1.0 1.0 1.0 1.0 0.2 1.0 1.0 0.2
neighbor 0.8 bin
timestep 0.002
thermo_style custom step temp etotal vol epair press pe
thermo 100
dump 1 all custom 100 dump.ellipse.gayberne &
id type x y z quatw quati quatj quatk
fix 1 all npt 2.6 2.6 0.5 xyz 0.0 8.0 0.5
compute_modify 1_temp extra ${dof}
run 100000
unfix 1
fix 2 all npt 2.6 2.6 0.5 xyz 8.0 8.0 0.5
compute_modify 2_temp extra ${dof}
run 100000
28 Ekim 2009 10:57 tarihinde ahmet yıldırım <ahmedo047@…24…> yazdı:
I don't know the answers to your questions. You will need
to carefully think about what the "temperature" of your system
is if you are deleting/adding degrees of freedom. And verify
that LAMMPS is computing a temperature consistent with
what those DOF are.
Steve
2009/10/28 ahmet yıldırım <[email protected]...>:
Dear steve,
I want them to translate but not rotate so I don’t integrate the rotational degrees of freedom.
I am using npt instead of npt/asphere for molecular alignment(as you said/suggest previously).I obtained the parallel orientation to the direction of z.
I entered Temperature=2.6 at initial file but temperature value didn’t reach to 2.6. Tempereture reachs to approximately 1.7. I think this problem is due to rotational degrees of freedom.Because I have to delete “command lines (dark lines as the following)” related to rotational degrees of freedom in input file. isn’t it?
You said And verify that LAMMPS is computing a temperature consistent with what those DOF are.
For molecular alignment(translate):
…
shape 1 1 1 3
set group all quat 0 0 1 0
compute rot all temp/asphere
###group spheroid type 1
###variable dof equal count(spheroid)+2
###compute_modify rot extra ${dof}
fix 1 all npt 3.0 3.0 0.5 xyz 0.0 8.0 0.5
###compute_modify 1_temp extra ${dof}
…
29 Ekim 2009 18:15 tarihinde Steve Plimpton <[email protected]> yazdı:
If you're constraining your ellipsoids to not rotate, I would treat
them with 3 DOF each. You'll need to figure out if that
is what you are doing.
Steve
2009/10/30 ahmet yıldırım <[email protected]...>: