Hi,
I have now upgraded the LAMMPS version and got rid of the 'illegal
create_atoms command' error. Valeria was right to point that out. Thanks to
everyone for your suggestions. Now I having a major problem, which I can't
make out why. It would be great if you could throw some light on this.
The Ag core of 441 atoms (radius 3 lattice units) equilibrated quickly. But
then when I introduced single silver atom at radius 6 lattice units and gave
it a small velocity toward the core, the temperature (and thus kinetic energy)
started going down rapidly when the single Ag atom came closer to the surface
of the core and entered its subsurface. The temperature went to zero when run
for longer time. There is just the Ag core and one Ag atom approaching it,
with vacuum in between. Could anyone give me a hint why the system is losing
energy. I have pasted my input file here.
Your help would be much appreciated.
I don't see anything obvious wrong. You might try removing the angular
momentum fix - I doubt it is doing much. You can zero the angmom
when velocities are created. I would run a smaller, shorter simulation
to debug it.
Steve
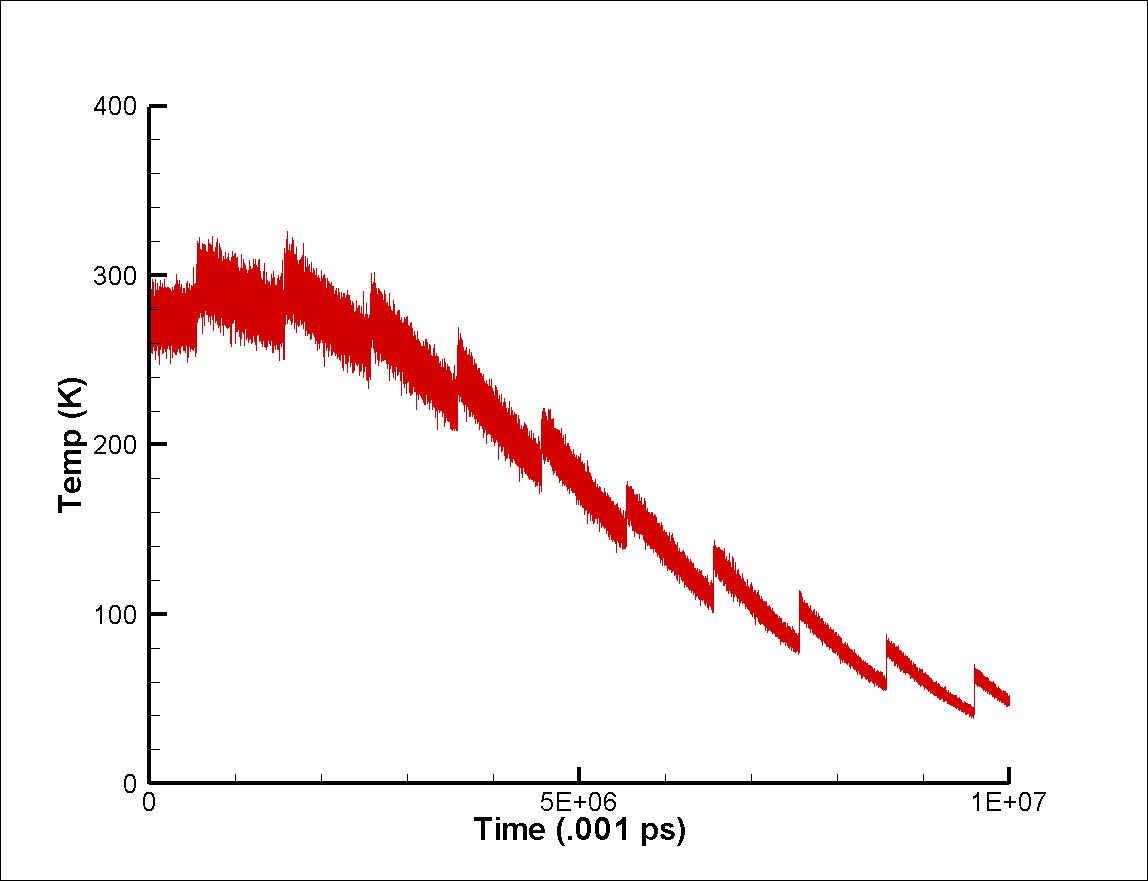
Steve and all group members,
Thanks for your suggestion. I ran another simulation. In this one, the core is
silver (441 atoms) and 10 singular silver adatoms are introduced one after
another at a gap of 1000 picoseconds. It is evident from the attached picture
that the temperature sharply goes up everytime when a new adatom (in this case
silver) is introduced and then steadily goes down. The total energy is also
dropping slowly. I have not changed the previous code other than incorporating
9 more silver adatoms and changing the delay between each.
I am still pondering over why this could happen. Any help would be vital for
my work and much appreciated.
Thanks,
Sourav
I don't see anything obvious wrong. You might try removing the angular
momentum fix - I doubt it is doing much. You can zero the angmom
when velocities are created. I would run a smaller, shorter simulation
to debug it.
Steve
> Hi,
>
> I have now upgraded the LAMMPS version and got rid of the 'illegal
> create_atoms command' error. Valeria was right to point that out. Thanks to
> everyone for your suggestions. Now I having a major problem, which I can't
> make out why. It would be great if you could throw some light on this.
>
> The Ag core of 441 atoms (radius 3 lattice units) equilibrated quickly. But
> then when I introduced single silver atom at radius 6 lattice units and
gave
> it a small velocity toward the core, the temperature (and thus kinetic
energy)
> started going down rapidly when the single Ag atom came closer to the
surface
> of the core and entered its subsurface. The temperature went to zero when
run
> for longer time. There is just the Ag core and one Ag atom approaching it,
> with vacuum in between. Could anyone give me a hint why the system is
losing
Thanks to Steve, Rutuparna and Wan Liang for your suggestions.
Reducing the timestep to 0.0001 and removing the fix momentum worked. Now I am
trying only removing the fix and keeping the timestep as before.
Could you please suggest one thing: if I am setting the angular momentum to be
zero with the following command
velocity all create 500.0 5812775 dist gaussian rot yes temp 1
will it still make sense to use the command
velocity all zero angular
immediately after it?
Thanks very much.
Sourav
If you need that small a timestep, your velocity is probably huge.
So you shouldn't delay the neighbor list build (i.e. use delay 1),
else you could lose energy due to missing pair interactions.
If you zero the angular momentum initially, you shouldn't need
to do it again, although if your new atom is off-axis and introduces
a large delta of angular momentum, then you might.
Steve